色素性视网膜炎查看源代码讨论查看历史

色素性视网膜炎RP是Retinitis Pigmentosa的简称是视网膜色素性变的英文缩写。视网膜色素变性是一种慢性、进行性、遗传性,营养不良性视网膜退行性变。因为本病的预后不好,所以香港东方日报以“恐怖的视网膜色素病变”大标题和成因未明,无法医治,不宜觉察,遗传后代的小标题提醒人们注意视网膜色素变性的严重性。香港文汇报则以“视网膜眼疾勿作夜盲“的大标题和专家警告视网膜色素变性会不知不觉夺去视力的小标题下,警告人们要认识视网膜色素变性这种病对患者眼睛造成危害之大。[1]
目录
视网膜色素变性(RP)的含义
视网膜色素变性(RP)指的是影响视网膜、引起视网膜功能退化的一种疾病。北京武警第二医院眼科主任医师杨春华主任、张振义主任经过临床近三十年的反复实践,采取"海勒氏动脉环,涡状静脉侧枝循环建立术治疗视网膜色素变性,有效率达95.6%。经北京大学视觉研究中心博士生导师蔡浩然教授手术前后眼电生理对比检测证实疗效显著。海勒氏动脉环涡状静脉侧枝循环建立术,是根据视网膜色素变性的病理变化,通过手术使脉络膜外层大血管、视神经软脑膜血管网、视乳头筛板前区和筛板区、视网膜扇形区的循环代谢得到明显改善和视功能逆转。早期(视网膜电图未熄灭型)手术后可以大幅度提高视力,拓宽视野,视网电图逐渐恢复正常。中晚期患者(视网膜图呈熄灭型,电脑视野残存10度以上,中心视力0.1以上),手术后视力略有提高,视野有所改善。
视网膜电图无改变,术后一年复诊,眼电图光峰暗谷阿登比有所改善。由杨春华主任、张振义主任领衔成立的专项课题组,经过27年的潜心研究、不懈探索,针对视网膜色素变性与脉络膜微循环异常密切相关这一重要中间环节,进行手术治疗干预。完善了"自体血管取材侧支循环建立术"治疗视网膜色素变性的学术论点。手术设计不损伤原有视力及视野,是现阶段可供选择的安全有效的手术治疗方式。在基础理论研究及临床实践过程中,得到北京大学医学部博士生导师,生物电学专家蔡浩然教授的支持,通过对患者手术前后严格的视觉生理:视网膜电图(F-ERG)、眼电图(EOG)、视觉诱发电位(P-VEP)各参数对比,充分论证了此手术对视网膜色素变性患者临床治疗的有效性。经卫生部医药卫生信息查新结论:能引起视网膜色素变性患者视觉电生理改善的治疗方式,国内外未见相同文献报道。证实了此项特色医疗服务项目在国内外眼科医疗领域处于领先水平。[2]
视网膜色素变性是眼科中的一种疑难疾病,被称为不治之症,是原发于视网膜营养不良的一种遗传性慢性眼病,多累及双眼,国内患者约30万,全世界约300万,其临床特点表现为早期夜盲、视野向心性缩小,最终呈管状视野,双目失明或频临失明,在失明眼病中占有相当大的比例。视网膜色素变性(RP)属于遗传性视杆、视锥细胞营养不良性疾病。以夜盲、视野缩小、眼底骨细胞样色素沉着为特征。数十年来国内外眼科界多项基础研究证实:"视网膜色素变性与脉络膜血流减少有关",夜盲、管状视野的形成预示脉络膜血流已无法满足视杆细胞的代谢需求,与脉络膜毛细血管区域血流循环功能下降相对应。我中心经过对中国、日本、美国、德国、哈萨克斯坦、缅甸、蒙古、菲律宾、加拿大等国家3000余例患者实施"自体血管取材侧支循环建立术"后证实,早、中期患者通过手术治疗可以不同程度改善视力、拓宽视野,眼底改善总有效率达96%;晚期患者能有效阻止病情进展,延缓失明。基础医学推动临床医学进展。手术临床实践验证了此手术设计与国内外眼微循环学说研究成果相吻合。 视风膜色素变性是原发于网脉络膜营养不良,以视网膜色素上皮、视网膜功能障碍为主要临床表现的一种先天遗传性致盲眼病。我国群体发病率大约1/4000,有30多万,全世界患者300多万。临床表现为早期夜盲,视野进行性缩窄,严重者呈管状,最终中心视力下降逐渐失明或频临失明。
患者一经确诊此病(不治之症)的结论给病人造成极大的心理损害,同时也给幸福的家庭蒙上一层阴影,辗转治疗,漫长的求医生涯令人心力交瘁,最终却摆脱不了失明的厄运。此项目经历了长达十八年的反复临床实践所取得显著疗效,通过手术前后眼电生理对比检测得以证实。经国医学科学院医学信息研究所检索,结论:能引起眼电生理改善的治疗方法未见相同国内、外文献报道。视网膜色素变性虽属遗传性疾病,但视网膜、脉络膜微循环障碍在本病的发生、发展过程中起到了至关重要的作用。祖国医学称本病为"高风内障"、"高风雀目",系先天禀赋不足。70年代由苏联眼科专家提出改善脉络膜微循环治疗此病;90年英国眼科学者证实:视网膜色素变性与脉络膜血流减少有关;此理论99年再次被北京同仁医院杨文利等采用多普勒检测而证实。根据视网膜色素变性病理改变,该中心采用建立侧枝循环手术治疗,使脉络膜外层大血管、视网膜扇行区的循环代谢得到了明显改善,视功能逆转。为医治视网膜色素变性患者闯出一条新路。
由于术式的不断改进,辅助治疗的逐渐完善,早期患者术后可提高视力,拓宽视野,视网膜电图逐渐恢复正常或接受正常;中期患者术后可提高视力,视野、视网膜电图不能改变,但60%患者术后一年眼电图光峰、暗谷、Arden比值有所改善或接近正常;晚期患者术后可有效阻止病情发展。中心的专家忠告视网膜色素变性患者一经明确诊断,应严密关注视网膜电图、电脑视野图形变化,千万不可延误治疗造成不可逆转的视功能损害。到目前为止,已成功治疗病人达550例,均收到良好效果,其中对26例患者进行5-15年远期观察,远期疗效明显优于近期疗效,未发现并发症。 视网膜色素变性患者一经确诊,即应严密关注视网膜电图、电脑视野图形及眼底变化,并在早、中期及时手术,避免贻误治疗时机,造成不可逆转的视功能损害,从而争取更好疗效。我中心全体医务人员将会通过不懈的努力,为视网膜色素变性患者提供尽可能良好的医疗服务。
视网膜色素变性是当今造成失明的重症眼病之一,美国眼科界统计资料显示,20--40岁年龄组,视网膜色素变性是主要的致盲眼病。据粗略的统计,全世界现有视网膜色素变性患者约150万。这个惊人的数字,并非逐年消减,而是与日俱增,难道这还不足以引起世人的关注吗!?视网膜色素变性早期,只有夜盲的症状,完全不影响工作,学习和正常生活,而病是在不知不觉的情况下对视力加以破坏的。尤其是处在幼儿时的孩子,想早期发现就更加困难了。视网膜结构精密,功能复杂,用肉眼是无法窥见的眼球后节的球内组织。1851年Helmhotz发明检眼镜,视网膜色素变性的眼底改变才被揭示。尽管如此,治疗仍是空谈。到本世纪70年代,视网膜色素变性才引起西方发达国家的注目。我国发现本病是在清朝早期,当时称之为"雀目内障"。顾名思义,恰如其分。病名命的好,况且也道出了辩证施治的依据,如:以健脾益气为主,加疏肝活血方剂,配合针灸、推拿、气功和滋补类药品等,有的方剂延续至今仍在使用。[3]
医学遗传学是人类遗传学的主要组成部分,是遗传学和医学结合的一门边缘科学,通过探讨人类疾病发生发展与遗传因素的关系,来提供诊断,治疗,预防遗传性疾病的科学依据,从而为改善热口素质做出贡献。随着医学的不断发展,人们生活水平不断提高,多数传染病得到控制,个别已经消失,一些慢性非感染疾病逐渐增多,成为现代医学突出的问题,迄今为止,人们已认识到遗传性疾病已超过了5000种,构成了对人类生命和健康的极大威胁。然而,伴随着现代高新技术的快速发展,基础医学尤其是免疫学及分子生物学和遗传学的研究也朝着深度和广度挺进,其中从细胞水平和分子水平进行研究的细胞遗传和分子遗传学发展的更是迅速,使得一些遗传性疾病的基因诊断和基因治疗更为可能。
如:从病人体内取出细胞经基因改造后,再重新注入体内,人为的改变异常基因,达到预防与治疗遗传病的目的;美国波士顿大学一研究机构用注射一种控制血管内皮生长素分泌的基因,该基因能指示人体生长出新的血管。对分子遗传学研究的成绩和操纵人类基因组合方面获得的成果,使应用重组DNA技术来治疗疾病成为可能,尽管这种基因工程目前还处于探索和实验阶段,但是已可望在数年内将人类染色体上10万个基因DNA顺序弄清,绘制出完整的人类基因组图谱--"生命的蓝图。"相信他这是一次具有方向性和本质性的措施。伴随着免疫遗传学和外科手术学的进展,对一些难以治疗的遗传性疾病,比如对视网膜色素变性的治疗,其前景是令人鼓舞的。
法国大诗人歌德说:"我要做的事,不过是伸手去收割旁人替我播种的庄稼而已。"而我所做的,则是把别人收获的"果实"分类理顺,再奉献给读者。由于我们水平有限,经验少,手头资料匮乏,错误之处定会很多,敬请眼科同道和读者指正,我们在此表示感谢。我们建立这个科研网站的目的,是呼吁社会各界关注视网膜色素变性这种遗传病,尽可能更多给予该病患者支持和帮助,唤起人们对视网膜色素变性病变的认识。同时呼吁眼科同道,开展科研协作,进行网站交流,让更多的RP患者病情得到有效控制和满意的治疗。如果视网膜色素变性的患者,能从我们这个网站里得到的一些启示,鼓起生活的勇气,认识到五彩缤纷的世界不会离你而去,那我们会感到无比欣慰。文献中有试用血管扩张剂、维生素A及B1、组织疗法、各种激素、中草药、针灸等方法,或可避免视功能迅速恶化。
视网膜

- 视网膜是位于眼球内部的精密的薄层组织,它包含着多层被称作“感光器”的感光细胞,这些感光细胞通过视神经与大脑相连。如果你把眼睛视作接受图像的照相机,那么视网膜就是记录图像的胶卷。
- 视网膜下面是视网膜色素上皮(RIPE)。视网膜色素上皮支持着视网膜感光细胞的工作。
- 视网膜上有两种感光细胞: 视锥细胞和视杆细胞。
- 视锥细胞集中在视网膜的中心部位 (称为黄斑区),负责中心视觉和分辨五颜六色。
- 视杆细胞分布在黄斑区外围,负责周边视觉和暗视觉。
- 视锥细胞和视杆细胞都将光线刺激转化为电脉冲,通过神经细胞传递到视神经, 经视神经再将信号传递至大脑,从而“看到”实际影像。
- RP患者的感光细胞逐渐变性,最终丧失功能。
原发性视网膜色素变性
原发性视网膜色素变性(primary pigmentary degeneratio of the retina)在历史上曾称为色素性视网膜炎(retinitis pigmentosa)。是一种比较常见的毯层-视网膜变性。根据我国部分地区调查资料,群体患病率约为1/3500。视网膜色素变性是一种少见的遗传性眼病。本病表现为慢性、进行性视网膜变性,最终可导致失明。
部分患者视网膜色素变性为显性遗传,父母双方只要有一方带致病基因,子女就会发病。也有部分患者视网膜色素变性为连锁性遗传,仅仅母亲带致病基因,子女才会发病。另有些病例同时伴有听力减退,这种类型视网膜色素变性多见于男性。
视网膜的一些感光细胞(视杆细胞)负责暗光下的视力。若视杆细胞逐渐变性,患者在暗光环境下视力明显减退(夜盲)。夜盲症状常在儿童期即出现,随时间发展,可出现进行性周边视野缺失。在晚期病例中,可仅残存一个小的中心视野(管状视野)和很窄的周边视野。
通过检眼镜检查,医生可发现视网膜上有某些具有诊断价值的特殊变化。也有数项检验可帮助进一步诊断。对家庭成员的检查可建立遗传模式。[4]
病因学

本病为遗传性疾病。其遗传方式有常染色体隐性、显性与性连锁隐性三种。以常染色体隐性遗传最多;显性次之;性连锁隐性遗传最小。目前认为常染色体显性遗传型至少有两个基因座位,位于第一号染色体短臂与第三号染色体长臂。性连锁遗传基因位于X染色体短壁一区一带及二区一带。 关于发病机制,近20~30年中,有了一些瓣的线索。根据电镜、组织化学、电生理、眼底血管荧光造影等检查资料推测,认为本病的发生,主要由于视网膜色素上皮细胞对视细胞外节盘膜的吞噬、消化功能衰退,致使盘膜崩解物残留、规程形成一层障碍物,妨碍营养物质从脉络膜到视网膜的转动,从而引起视细胞的进行性营养不良及逐渐变性和消失。这个过程已在一种有原发性视网膜色素性的RCS鼠视网膜中得到证实。至于色素上皮细胞吞噬消化功能衰竭的原因,目前还不清楚。可能与基因异常,某种或某些酶的缺乏有关。
在免疫学方面,近年研究发现本病患者体液免疫、细胞免疫均有异常,玻璃体内有激活的T细胞、B细胞与巨噬细胞,视网膜色素上皮细胞表达HLA-DR抗原,正常人则无此种表现。同时也发现本病患者有自身免疫现象,但对本病是否有自身免疫病尚无足够依据。在生化方面,同时也发现本病患者有自身免疫现象,但对本病是否有自身免疫病尚无足够依据。在生化方面,发现本病患者脂质代谢异常,视网膜中有脂褐质的颗粒积聚;锌、铜、硒等微量元素及酶代谢亦有异常。综上所述,本病可能存在着多种不同的发病机理。
病理改变
临床得到的标本均为晚期病例。光学显微镜下所见的主要改变为视网膜神经上皮层、特别杆细胞的进行性退变,继以视网膜由外向内各层组织的逐渐萎缩,伴发神经胶质增生。色素上皮层也发生变性和增生,可见色素脱失或积聚,并向视网膜内层迁徙。视网膜血管壁发生玻璃样变性而增厚,甚至管腔完全闭塞。脉络膜血管可有不同程度硬化,毛细血管完全或部分消失。视神经可完全萎缩,视肋上常有神经胶质增生,形成膜块,与视网膜内的胶质膜相连接。检眼镜下所见视盘的蜡黄色,一般认为与此有关。
临床表现
症状与功能改变
- 夜盲:为本病最早出现的症状,常始于儿童或青少年时期,且多发生在眼底有可见改变之前。开始时轻,随年龄增生逐渐加重。极少数患者早期亦可无夜盲主诉。⑵暗适应检查:早期锥细胞功能尚正常,杆细胞功能下降,使杆细胞曲线终未阈值升高,造成光色间差缩小。晚期杆细胞功能丧失,锥细胞阈值亦升高,形成高位的单相曲线。
- 视野与中心视力:早期有环形暗点,位置与赤道部病变相符。其后环形暗点向中心和周边慢慢扩大而成管状视野。中心视力早期正常或接近正常,随病程发展而逐渐减退,终于完全失明。
- 视觉电生理:ERG无反应,尤其b波消失是本病的典型改变,其改变常早于眼底出现改变。EOG LP/DT明显降低或熄灭,即使在早期,当视野、暗适应、甚至ERG等改变尚不明显时,已可查出。故EOG对本病诊断比ERG更为灵敏。
- 色觉:多数患者童年时色觉正常,其后渐显异常。典型改变为蓝色盲,红绿色觉障碍较少。
眼底检查所见典型的改变

本病早期虽已有夜盲,眼底可完全正常。俟后随病程进展而渐次出现眼底改变。典型的改变有:
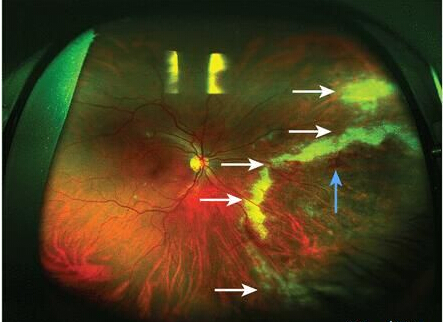
- 视网膜色素沉着:始于赤道部,色素呈有突的小点,继而增多变大,呈骨细胞样,有时呈不规则的线条状,围绕赤道部成宽窄不等的环状排列。色素多位于视网膜血管附近,特别多见于静脉的前面,可遮盖部分血管,或沿血管分布,于血管分支处更为密集。以后,色素沉着自赤道部向后极和周边逐渐扩展,最后布满整个眼底。在此同时,视网膜色素上皮层色素脱失,暴露出脉络膜血管而呈豹纹状眼底。晚期脉络膜血管亦硬化,呈黄白色条纹。玻璃体一般清晰,有时偶见少数点状或线状混浊。
- 视网膜血管改变:血管一致性狭窄,随病程进展而加重,尤以动脉为显著。在晚期,动脉成细线状,于离开视盘一段距离后即难以辨认而似消失,但不变从白线,亦无白鞘包绕。
- 荧光血管眼底造影所见:背景荧光大片无荧光区,提示脉络膜毛细血管层萎缩。视网膜血管可有闭塞,有时还可见到后极部或周边部斑驳状荧光斑。
特殊临床类型
- 单眼性原发性视网膜色素变性:非常少见。诊断为本型者,必须是一眼具有原发性视网膜色素变性的典型改变,而另眼完全正常(包括电生理检查),经五年以上随访仍未发病,才能确定。此型患者多在中年发病,一般无家族史。
- 象限性原发性视网膜色素变性:亦甚少见。特点为病变仅累及双眼同一象限,与正常区域分界清楚。有相应的视野改变,视力较好,ERG为低波。荧光造影显示病变区比检眼镜下所见范围大。本型常为散发性,但也常染色体显性、隐性与性连锁隐性遗传的报告。
- 中心性或旁中心性原发性视网膜色素变性:亦称逆性进行性视网膜色素变性。初起即有视力减退与色觉障碍。眼底检查可见黄斑部萎缩病变,有骨细胞样色素堆积,ERG呈低波或不能记录。早期以锥细胞损害为主,后期才有杆细胞损害。晚期累及周边部视网膜,并出现血管改变。
- 无色素性视网膜色素变性:是一种有典型视网膜色素变性的各种症状和视功能的检查所见。检眼镜下亦有整个眼底灰暗、视网膜血管变细、晚期视盘蜡黄色萎缩等改变,无色素沉着,或仅在周边眼底出现少数几个骨细胞样色素斑,故称为无色素性视网膜色素变性。有人认为本型是色素变性的早期表现,病情发展后仍会出现典型的色素。因此不能构成一单独临床类型。但亦确有始终无色素改变者。本型遗传方式与典型的色素变性相同,有显性、隐性、性连锁隐性遗传三型。
鉴别诊断

根据上述病史、症状、视功能及检眼镜检查所见,诊断并无太大困难。但当与一些先天生或后天性脉络膜视网膜炎症后的继发性视网膜色素变性注意鉴别。
先天性梅毒和孕妇在妊娠第3个月患风诊后引起的胎儿眼底病变,出生后眼底所见与本病几乎完全相同,ERG、视野等视功能检查结果也难以区分。只有在确定患儿父母血清梅毒反应阴性及母亲妊娠早期无风疹病史后,才能诊断为原发性色素变性。必要时还需较长时间随访观察,先天性继发性色素变性在出生时即已存在,病情静止。
后天性梅毒和某些急性传染病如天花、麻疹、猩红热、流行性腮腺炎等,均可发生脉络膜视网膜炎,炎症消退后的眼底改变,有时与原发性色素变性类似。当从病史、血清学检查以及眼底色素斑大而位置较深、形成不规则(非骨细胞样)、有脉络膜视网膜萎缩斑、视盘萎缩呈灰白色不是蜡黄色、夜盲程度较轻等方面加以鉴别。
治疗措施
文献中有试用血管扩张剂、维生素A及B1、组织疗法、各种激素、中草药、针灸等方法,或可避免视功能迅速恶化。
- 遮光眼镜片之选用 强光可加速视细胞外节变性,所以必须戴用遮光眼镜。镜片的颜色从理论上说,应采用与视红同色调的红紫色,但有碍美容用灰色,阴天或室内用0~1号;晴天或强光下用2~3号灰色镜片。深黑色墨镜并不相宜。绿色镜片禁用。
- 避免精神和肉体的过度紧张 过度紧张时体液内儿茶酚胺(catecholamine)增加,脉络膜血管因此收缩而处于低氧(hypoxia)状态,使视细胞变性加剧。我国传统的气功(静功),能以自己的意志高速大脑皮层及机体各器官的活动,如持之以恒,对防止本病视功能迅速恶化方面可能有益。
- 补充叶黄素:叶黄素广泛分布在正常人视网膜的一种具有视网膜保护作用的物质,是视网膜组织的重要组成成分,叶黄素具有优秀的搞氧化、对抗光损伤的能力以及视细胞营养作用,能够防止氧自由基及有害光对视网膜视细胞及色素上皮细胞的损害,提高视细胞的活性。人体自身不能合成叶黄素,主要来源于外界摄取,一旦缺乏,就会产生各种视网膜疾病,几乎所有视网膜病变都与叶黄素的缺乏有关。
并发症

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
后极性白内障是本病常见的并发症。一般发生于晚期、晶体混浊呈星形,位于后囊下皮质内,进展缓慢,最后可致整个晶体混浊。约1%~3%病例并发青光眼,多为宽角,闭角性少见。有人从统计学角度研究,认为青光眼是与本病伴发而非并发症。约有50%的病例伴有近视。近视多见于常染色体隐性及性连锁性隐性遗传患者。亦可见于家族中其他成员。聋哑病兼患本病者亦高达19.4%。视网膜与内耳Corti器官均源于神经上皮,所以二者的进行性变性可能来自同一基因。
色素变性与耳聋不仅可发生于同一患者,也可分别发生于同一家族的不同成员,但二者似乎不是源于不同基因,可能为同一基因具有多向性所致。本病可伴发其他遗传性疾病,比较常见者为间脑垂体区及视网膜同时罹害的Laurence-Moon-Bardt-Biedl综合征。典型者具有视网膜色素变性、生殖器官发育不良、肥胖、多指(趾)及智能缺陷五个组成部分。该综合征出现于发育早期,在10岁左右(或更早)已有显著临床表现,五个组成部分不是具备者,称不完全型。此外,本病尚有一睦眼或其他器官的并发或伴发疾病,少见。
预后
本病隐性遗传患者发病早、病情重,发展快,预后极为恶劣。以30岁左右时视功能已高度不良,至50岁左右接近全盲。显性遗传患者则反之,偶尔亦有发展至一定程度后趋于静止者,故预后相对地优于隐性遗传型。因此可等到勉强正常就学和就业的机会。本病隐性遗传者,其先辈多有近亲联烟史,禁止近亲联烟可使本病减少发生约22%。另外,隐性遗传患者应尽量避免与本病家族史者结婚,更不能与也患有本病者结婚。显性遗传患者,其子女发生本病的风险为50%。
视频
视频