混合性結締組織病

混合性結締組織病(mixed connective tissue disease,MCTD)是一種綜合徵,其特點為臨床上具有系統性紅斑狼瘡、多發性肌炎及系統性硬化症等結締組織病的臨床表現,但又不符合其中一種疾病的診斷,且在血清中有高效價抗核糖核蛋白(RNP)抗體的一種自身性免疫性疾病。目前許多學者認為,MCTD只不過是某種風濕性疾病的中間過程或亞型。隨訪結果發現MCTD可發展成系統性紅斑狼瘡或硬皮病或其他風濕性疾病。因此,MCTD可以認為是一有特色的未分化的風濕性疾病。[1]
目錄
[隱藏]症狀體徵
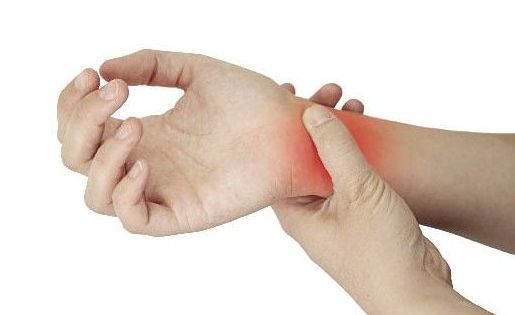
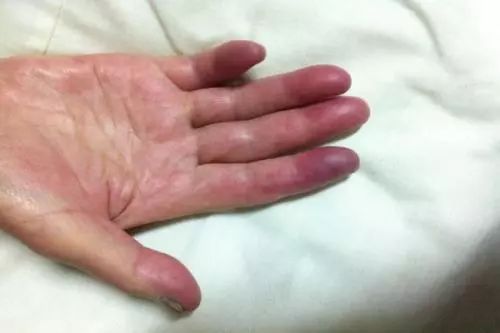
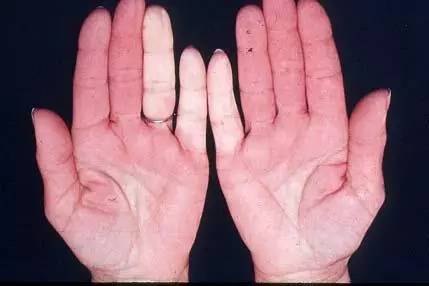
MCTD最常見的臨床表現是多關節炎、雷諾現象、肺部受累、手腫脹、指(趾)皮膚硬化、肌炎以及食管功能障礙,脫髮、皮疹、淋巴結病、肝脾腫大、漿膜炎、心、腎及中樞神經系統損害是較少見的表現(表1)。
關節炎的主要表現為關節腫痛,嚴重者有關節潰爛、畸形,但皮下結節少見。皮膚硬化多見於指(趾)末端和面部,很少廣泛累及。臟器受累主要有肺、心血管、腎臟以及神經系統。肺部受累可表現為瀰漫性間質浸潤和肺彌散功能障礙,可有肺動脈高壓。心臟損害最常見的表現是心包炎,見於1/3病人,其次為二尖瓣脫垂、心室擴大、心律失常等。神經精神症狀也不少見。半數病人有血液系統受損,最常見的症狀是出血,血小板和白細胞減少偶見,脾臟腫大見於20%病人。MCTD的腎臟病變少見。一旦出現腎臟受累,常可表現為無症狀蛋白尿、血尿、腎病綜合徵或不同程度腎功能不全,個別患者可完全無症狀。出現下列情況提示MCTD合併腎損害:
- 抗RNP抗體升高。
- 血清補體下降和(或)抗雙鏈DNA(抗ds-DNA)抗體滴度升高。
- 抗核抗體(ANA)陽性 除去腎血管性高血壓外,MCTD腎損害極少導致死亡,而心肺疾病是死亡的首要原因。
病理病因
MCTD病因未明。其發病可能與免疫紊亂有關,有關證據為血清中持續存在高滴度抗RNP抗體,顯著多克隆高丙球蛋白血症,B淋巴細胞活性過高,抑制性T細胞缺陷,疾病活動期血循環免疫複合物增加,25%病人有低補體血症,發現血管壁、肌纖維膜、腎小球基底膜及皮膚表皮和真皮結合部位有IgG、IgM、IgA和補體沉積。本病與HLA之間的關係正在研究中,有人認為其有特定免疫遺傳學背景,與HLA-DR4、-DR5密切相關,認為MCTD為一獨立性疾病。[2]
發病機制
MCTD的發病機制尚不清楚。以下證據表明,其發病機制可能與免疫介導有關:循環免疫複合物在腎小球沉積,血清抗RNP抗體陽性,許多組織廣泛的淋巴細胞、漿細胞的浸潤,高γ-球蛋白血症及T細胞抑制。
抗RNP抗體的抗原位點是核RNA蛋白複合物,低分子量小核糖核酸蛋白(SnRNPs)參與核RNA合成。MCTD患者血清中除SnRNPs抗體外,還有高分子量與核基質連接的其他抗體。已經證實IgG抗RNP抗體通過Fc受體與細胞內部連接,但通過何種途徑激活免疫反應仍不清楚。
SLE患者的血清抗體與MCTD不同,兩者異常T淋巴細胞亞群也各不相同。MCTD免疫複合物數量增加可能與網狀內皮系統免疫複合物清除受損有關。[3]
遺傳因素可能與MCTD有關,但是MCTD和SLE有不同的家族發病體系。






疾病診斷
重疊綜合徵(overlapping syndrome)
是指同一個患者同時或先後患有明確2種或2種以上結締組織病的重疊,因此與MCTD明顯差別。儘管MCTD臨床上有多種病的重疊症狀,但它有其自身的診斷標準和特點。
重疊綜合徵臨床表現為同一時期內並存兩種以上風濕性疾病,如系統性紅斑狼瘡與結節性多動脈炎並存,系統性紅斑狼瘡與系統性硬化症並存,幼年型類風濕性關節炎除有嚴重關節炎和皮下小結外,還有抗核抗體陽性和血管炎,即與系統性紅斑狼瘡並存。此外,系統性硬化症也可出現抗核抗體陽性、嚴重關節炎、白細胞減少、溶血性貧血等。[4]
重疊綜合徵也可先後出現兩種以上風濕性疾病,如患皮肌炎多年後,又出現雙手指關節炎,可視為皮肌炎與類風濕關節炎重疊。系統性紅斑狼瘡的病程進展過程中,又出現了典型的類風濕關節炎的關節畸形,可診斷為系統性紅斑狼瘡和類風濕關節炎的重疊。
重疊綜合徵的治療較治療單純患某一種疾病困難。應用藥物應取決於以何種風濕性疾病為主。主要藥物為腎上腺皮質激素和免疫製劑,劑量和用法參照各有關風濕性疾病節。
本綜合徵的預後較單一的風濕性疾病要差。同時還取決於哪兩種病的重疊。與MCTD相比,MCTD的5年存活率為93%,而重疊綜合徵的存活率僅為53%。
系統性紅斑狼瘡、多發性肌炎、系統性硬化症 MCTD各具有三者中的某一些特徵、而又不能用某一種病解釋MCTD所有表現。
檢查方法

實驗室檢查
MCTD的實驗室特點。
- 一般檢查:部分患者有貧血、白細胞減少及血小板減少,血沉增快,Coombs直接試驗陽性。約3/4患者有高γ-球蛋白血症。活動期患者可有CPK、醛縮酶、乳酸脫氫酶及轉氨酶顯著升高。腎臟受累時可見蛋白尿、血尿、腎病綜合徵或不同程度腎功能不全的改變。
- 血清免疫學異常:MCTD以血清中高濃度抗RNP抗體(>1∶1000)為特點。抗RNP抗體在MCTD中檢出率高、滴度高,是MCTD特徵性抗體。近年來抗RNP抗體亦發現於SLE及其他結締組織病中,但檢出率及滴度均較低。幾乎所有MCTD均有血清ANA陽性,血清ANA陽性診斷MCTD不具有特異性。
少數患者可有抗sm抗體和抗ss抗體陽性,類風濕因子(RF)陽性。血清補體水平可正常、升高或降低,低補體血症與腎損害程度無相關性。
其他輔助檢查
- 病理:MCTD主要組織病理改變是廣泛的增殖性血管病變,包括動脈和小動脈內膜的增殖和中層肥厚,導致大中血管如主動脈、肺動脈、腎動脈、冠狀動脈和許多臟器小血管的狹窄,但無明顯炎症細胞浸潤。5%~25%有腎臟受累,兒童較成年人多見。受累腎臟的病理變化也具有混合病變的特點,腎小球、腎血管及腎間質均可出現病變。腎小球病變表現為細胞增生,局灶性基底膜增厚,繫膜增生,可有繫膜增生型、局灶增生型、膜型、膜增殖型腎小球腎炎的表現;有小動脈顯著內膜增生及閉塞,有血管炎病變;腎間質常見淋巴細胞、單核細胞和漿細胞大片浸潤。
- 腎活檢光鏡特點:MCTD腎損害多為輕微病變腎小球腎炎,也可以是嚴重增生性病變,可自然好轉或轉為腎功能衰竭。根據其病理特點,分為免疫複合物介導腎小球腎炎和血管硬化性或硬皮病樣腎損害。MCTD腎活檢標本光鏡特點見表3,4,成人和兒童均以繫膜性腎小球腎炎和膜性腎小球腎炎最多見。
- 免疫複合物介導腎小球腎炎:免疫複合物介導腎小球腎炎類似狼瘡性腎炎,最常見的是繫膜增生性腎小球腎炎。其次是膜性腎小球腎炎,常伴有腎小球繫膜細胞增多、膜性缺損,間質免疫複合物沉積。瀰漫增生性腎小球腎炎、膜增生性腎小球腎炎、間質性腎小球腎炎伴腎小球基底膜免疫複合物沉積,也有報道。新月體形成較少見,但也可見於膜增生性腎小球腎炎和膜性腎小球腎炎。部分患者腎活檢標本中可見到微管病毒樣顆粒,但缺乏特異性。
- 硬皮病樣腎損:硬皮病樣腎損害較免疫複合物介導腎小球腎炎少見,與硬皮病的血管增生、硬化類似。免疫複合物介導腎小球腎炎也可伴有血管的增生、硬化。腎小血管和腎動脈內層、中層增厚多見於疾病晚期,常導致高血壓,治療較困難。
免疫熒光顯微鏡特點
約1/3患者皮膚活檢在表皮與真皮交界處可見IgG、IgM及或C3沉積。腎小球繫膜增生性腎炎可見IgG、補體在腎小球繫膜沉積,同樣的,膜性腎小球腎炎可見IgG、補體沿毛細血管壁沉積。免疫複合物易於沉積在腎小球,腎活檢標本中最常見的免疫反應物是IgG和IgM(31%)。約1/4病人有IgG、IgA和IgM沉積,與狼瘡性腎炎類似。23%病人與狼瘡性腎炎相反,只有IgG沉積,狼瘡性腎炎的免疫熒光表現多為「滿堂亮」(表5),C3是伴隨免疫球蛋白的常見補體成分。MCTD部分腎活檢標本在小管區可見核直接免疫熒光。
超微結構
MCTD腎損害患者電子緻密物最常沉積在上皮下(44%),其中約3/4活檢標本同時伴有腎小球繫膜區電子緻密物沉積,僅1/4標本僅見上皮下沉積,即「單純」膜性腎小球腎炎。腎小球繫膜未受累及在MCTD中較少見,不到1/5的患者可見電子緻密物的內皮下沉積,上皮下沉積總是伴隨內皮下沉積出現。45例活檢標本中3例(7%)在內皮下和腎小球繫膜區有電子緻密物沉積,提示MCTD的腎臟損害較狼瘡腎炎輕。
雖然MCTD的腎損害類似狼瘡性腎炎,但其病理分型多為輕微繫膜增生性或膜性。而在狼瘡腎炎,局灶性或瀰漫性增生占60%,腎小球繫膜增生占25%,膜性腎小球腎炎占10%。
併發症狀
可發生指、趾端缺血性潰瘍或壞死;少數並發胸膜炎,非間質纖維化,肺動脈高壓;可並發心包炎、心肌炎、心律失常、心瓣膜病變、心功能不全等;可並發腎功能衰竭,多發性神經炎,無菌性腦膜炎,癲癇等。尚有淋巴結腫大,少數肝脾腫大。
預後
MCTD為慢性風濕性疾病,病程較長,但經適當治療可獲臨床緩解。極少數嚴重病例可死於腎功能不全。成人死亡率為4%~7%,死亡原因為肺動脈高壓、腎功能不全、心肌炎、心肌梗死、心力衰竭、腦栓塞和泛發性血管炎、結腸穿孔。本病腎損害預後比狼瘡性腎炎好,死因主要為併發症及激素治療的不良反應等。






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
用藥治療
本病治療視其嚴重程度而定,臨床主要採用糖皮質激素治療。
- 輕型MCTD :可先用秋水仙鹼、非甾體抗炎藥或抗瘧藥治療,非甾體抗炎藥或15~30mg/d小劑量皮質激素治療即可控制病情。激素對以SLE為主要表現的效果好,而對以原發性舍格倫綜合徵(PSS)為主要表現的效果差。
- 重型MCTD:當MCTD伴有嚴重系統及器官損害表現與腎臟病變,如出現腎病綜合徵時,宜用中到大量的糖皮質激素治療,一般用潑尼松劑量為1mg/(kg·d),口服,治療數天至數周后症狀可顯著改善。此後,改為小劑量維持,每天或隔天用藥1次。注意緩慢撤藥,以防引起病情反覆,總療程可視其嚴重程度決定,一般需數年完成。當有嚴重的內臟病變,或以PSS為主要表現,對糖皮質激素反應較差時,可加用環磷酰胺等免疫抑制劑。膜性腎小球腎炎對皮質激素治療無效,也可用細胞毒類藥物治療。對嚴重危及生命的病變如心包炎、中樞神經系統病變、腎病綜合徵和腎衰,必須用糖皮質激素維持治療。一般說來,MCTD腎損害對糖皮質激素治療敏感,而硬皮病療效差。雷諾現象嚴重者可加用鈣離子拮抗藥,或血管擴張藥妥拉唑啉、丹參製劑等,可有一定效果。高血壓危象時應積極儘早控制血壓,使腎功能得以改善,目前首選藥物為血管緊張素轉換酶抑制劑。
飲食保健
- 混合性結締組織病吃哪些食物對身體好:高蛋白、高熱量、易消化的食物,雞鴨魚肉,五穀雜糧,蔬菜瓜果均不可忽視,應搭配合理。
- 混合性結締組織病最好不要吃哪些食物:少食辛辣刺激及生冷、油膩之物。
預防護理
MCTD腎損害的預防,主要依賴於早期診斷和合理有效治療。本病死因主要是感染等併發症及激素治療的不良反應,極少數嚴重病例可死於腎功能衰竭和中樞神經系統病變,故對本病的早期診斷和合理有效治療可預防併發症的發生和延長存活期。
視頻