亨廷頓病檢視原始碼討論檢視歷史

{kind=link}

{kind=link}

{kind=link}
亨廷頓病(Huntington disease,HD)是一種以不自主運動、精神異常和進行性痴呆為主要臨床特點的顯性遺傳性神經系統變性病。屬於基因動態突變病或多谷酰胺重複病的範疇。因亨廷頓病以舞蹈症狀為突出的臨床症狀,曾將本病命名為大舞蹈病、亨廷頓舞蹈病、慢性進行性舞蹈病或遺傳性舞蹈病。
1872年由美國內科醫師Huntington對亨廷頓病的臨床症狀首先進行了描述,1911年Alzheimer對病理改變作了觀察,1993年確定其致病基因位於第4對常染色體短臂63位點,此基因編碼的蛋白,命名為亨廷素(Huntingtin)。病理改變特點是紋狀體和大腦皮質的神經細胞脫失,最近發現在大腦皮質存在泛素陽性神經細胞核內包涵體和營養不良神經突起。
亨廷頓病患者多數發病年齡在25~40 歲,平均發病年齡在40 歲,此病持續5~30 年,平均14 年。5%~10%的患者發病年齡在10~20 歲,1%的患者發病年齡在兒童期,個別患者的發病年齡在80 歲以後。目前沒有任何藥物可以改變亨廷頓病的自然病程,但可以採取措施改善臨床症狀、減少舞蹈樣動作。治療集中在對心理與神經徵候兩方面的症狀治療,同時進行必要的支持治療。
病因
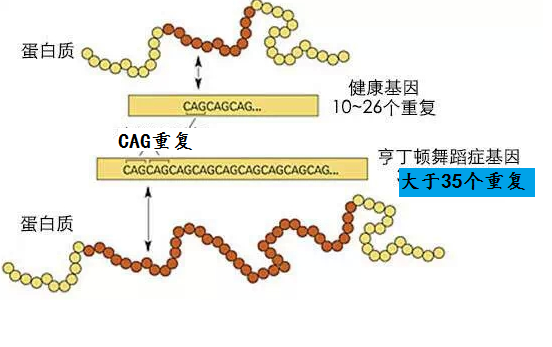
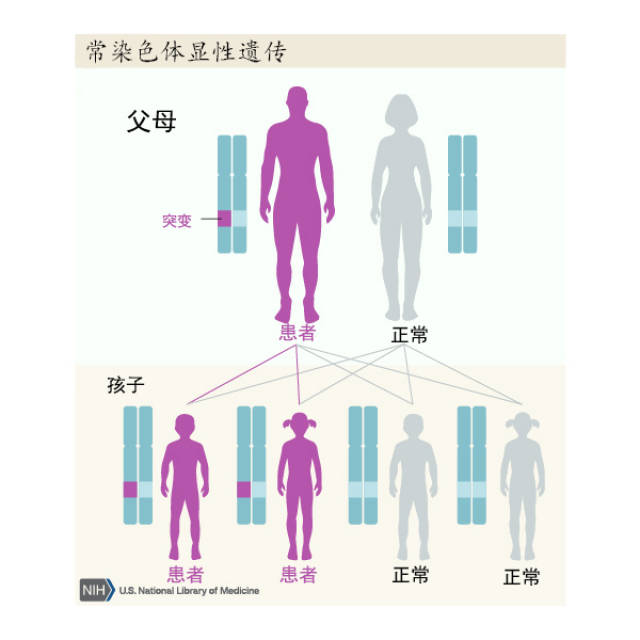
亨廷頓病是影響紋狀體和大腦皮質的常染色體顯性遺傳病,呈完全外顯率,受累個體後代50%發病。HD為4號染色體短臂4p16.3的Huntingtin基因突變所致,基因產物為CAG三核苷酸重複擴增產生Huntingtin蛋白,正常人為11~34個CAG重複序列,HD為40個以上。只要了遺傳致病基因,或早或晚會出現症狀,純合子與雜合子的臨床症狀無明顯差異,臨床亦偶見散發病例。根據發病年齡,HD可分為青年型(20歲前發病)及成年型。
發病機制及病理生理
如果把大腦想成是一顆球,那麼在靠近球心的部位 (也就是大腦的深部),有個由眾多神經細胞組成的構造叫基底核 (basal ganglia)。它與大腦的運動、情緒、學習、記憶功能有密不可分的關係。就運動功能而言,基底核扮演着大腦秘書的角色,將大腦發出的命令先行處理、修飾過,再把這個命令傳回大腦負責運動的部位,然後才把指令傳送到身體各處肌肉,指揮我們的一舉一動。[1]
亨廷頓病患者體內的異常亨廷頓蛋白首先會影響其腦內的基底核,使得基底核無法修飾或抑制大腦的指令,於是全身肌肉便不受控制地運動,表現為舞蹈樣動作。到了疾病的晚期,連負責下達指令的大腦表層也會逐漸死亡,屆時病人可能失去所有行動能力,並出現認知功能下降甚至痴呆。*[2]
亨廷頓病患者主要病理改變為基底節區萎縮,其中以尾狀核最為明顯,殼核和蒼白球也有不同程度的萎縮。神經元缺失主要見於基底節區,其中尾狀核和殼核的神經元功能障礙與舞蹈樣動作有關,皮質神經元缺失可能與痴呆有關。[3]
亨廷頓病的臨床表現
亨廷頓病為常染色體顯性遺傳。子女的發病幾率是50%。父系遺傳占優勢者發病較早,而母系遺傳占優勢者發病較晚。但如母親已發病,在妊娠過程中,由於母體與胎兒的相互作用,大部分胎兒流產。而由父系遺傳的小孩多能存活。和其他多谷酰胺重複病一樣,亨廷頓病的遺傳呈現遺傳早發現象,即一代比一代發病早,且一代比一代症狀重。
亨廷頓病的臨床症狀包括三方面,即運動障礙、認知障礙和精神障礙,這些臨床表現均可以作為首發症狀出現。
1、 運動障礙
進行性發展的運動障礙表現為四肢、面、軀幹的突然、快速的跳動或抽動,這些運動不可預先知道,也可以表現為不能控制的緩慢運動。查體發現舞蹈樣不自主運動和肌張力不全。舞蹈樣不自主運動是本病最突出特徵,大多開始表現為短暫的不能控制的裝鬼臉、點頭和手指屈伸運動,類似無痛性的抽搐,但較慢且非刻板式。隨病情發展,不隨意的運動進行性加重,出現典型的抬眉毛和頭屈曲,當注視物體時頭部跟着轉動,患者行走時出現不穩,騰越步態,加上不斷變換手的姿勢,全身動作像舞蹈。在疾病後期患者因全身不自主運動而不能站立和行走。即使坐着也不穩,身體扭動,突然站起又突然坐下,臥床後軀幹和肢體仍不停的扭動。當病情發展時,隨意運動受損愈益明顯,動作笨拙、遲緩、僵直,不能維持複雜的隨意運動,出現吞咽困難、講話吞吞吐吐和構音障礙。出現不正常的眼球活動異常。在病的晚期隨意運動減慢,呈現出四肢不能活動的木僵狀態。多數患者腱反射和感覺正常。
舞蹈樣運動障礙是成年型亨廷頓病的典型運動障礙。在20歲前起病的少年型患者(占亨廷頓病的5%~10%)中,以不動性肌強直為主要運動障礙。表現為肌強直、肌陣攣,至晚期則呈角弓反張。此外與成人患者不同,約50%的少年型亨廷頓病者有全身性癲癇發作。
2 認知障礙
進行性痴呆是亨廷頓病患者另一個特徵。痴呆在早期具有皮質下痴呆的特徵,後期表現為皮質和皮質下混合性痴呆。
認知障礙在亨廷頓病的早期即可出現。開始表現為日常生活和工作中的記憶和計算能力下降,患者記住新信息僅有輕度損害,但信息作修飾以便有效儲存有明顯困難,回憶也有顯著缺陷。由於詞的流利性、視空間功能及對社會和人際關係的判斷能力下降,病人變的比較混亂,出現人格的改變。
言語的改變,包括口語流利性測驗不良,輕度找詞困難和構音障礙。口語流利性損害是亨廷頓病最早能計量查出的認知功能不正常之一。在病的中期和晚期,患者不能完成需要組織、連續和語言學精心加工的語言測驗,也不能完成需回憶不常用詞的命名測試。但這些測試還需要記憶和認識能力,超出了語言範圍。沒有典型的錯語和失語症,但構音和韻律障礙為本病患者的突出特徵。舞蹈樣運動障礙常可累及舌和唇,破壞了發音的韻律和敏捷性,妨礙了言語的量、速度、節律和短語的長度,使口語呈現一種暴發性質。由於患者仍保留詞的識別記憶及對手的識別和對物的命名能力,亨廷頓病患者能繼續與人交流。 隨病情發展,集中力和判斷力進行性受損。患者缺乏啟動解決問題的行為。在需要計劃和連續安排信息的作業上感到特別困難。視空間能力下降,對結構的判斷有困難。在需要連續安排運動的額葉系統測驗上,如手的連續變換動作有困難。
3、 精神障礙
首先出現的精神狀態變化為人格行為改變,包括焦慮、緊張、興奮易怒、或悶悶不樂、或不整潔以及興趣減退,出現反社會行為、精神分裂症、偏執狂和幻覺。情感障礙是最多見的精神症狀,且多出現在運動障礙發生之前。由於情感障礙出現在患者的運動障礙出現之前,或了解其家族疾病特點之前,所以不是反應性障礙。此外抑鬱症狀的發生率也很高,對患者的重度抑鬱症狀如能早期發現並及時治療,可預防自殺。亨廷頓病患者的神經和精神性障礙進行性衰退,最後患者處於呆傻、緘默狀態。
4、青少年型Huntington舞蹈病
在兒童及青少年期起病,20歲前起病約10%,年齡小於4歲起病約5%。臨床表現與成人HD不同,病程進展較快,肌張力障礙是突出表現,常以強直替代舞蹈樣運動。尚可見Parkinson綜合徵、小腦性共濟失調、眼球運動異常、肌陣攣及癲癇發作等,可出現精神衰退及行為異常,部分患者表現運動過度。少數病例運動症狀不典型(Westphal變異型),表現進行性肌強直和運動減少,舞蹈-手足徐動樣症狀不明顯,多見於兒童期或20歲以前發病者。癲癇和小腦性共濟失調也是青少年型常見特點,伴痴呆和家族史可提示診斷。
輔助檢查
1、遺傳學檢測
是確診重要手段,PCR 法檢測亨廷頓基因(TT15)中CAG 重複拷貝數,正常人不超過38 個拷貝,患者在39 個以上,陽性率高,只需檢測患者本人,可作到疾病症狀前診斷和產前診斷等。
2、腦電圖
可有瀰漫性異常,無特異性。主要為低波幅快波,尤其額葉明顯,異常率占88.9%。α活動減少,波幅降低。視覺誘發電位波幅降低,但首波部分潛伏期正常。患者P100 不正常,檢測P300 可作為早期認知功能下降的客觀指標。
3、影像學檢查
頭部CT或MRI對於診斷亨廷頓病具有重要的臨床價值,典型的影像學特點是雙側尾狀核萎縮,導致側腦室額角外側面向外膨起。SPECT 檢查發現尾狀核和豆狀核區域血流明顯下降,額葉和頂葉血流也有下降,與患者這些部位的病理改變有關。PET 表現尾狀核區葡萄糖代謝明顯降低,尾狀核區的代謝活性下降可出現在尾狀核萎縮前。
臨床診斷標準
1.典型HD的家族史。
2.非其他因素導致的進行性運動異常伴舞蹈和僵直。
3.非其他因素導致的精神障礙伴隨進行性痴呆。
影像學檢查發現對稱性尾狀核萎縮可以進一步支持亨廷頓病的診斷。在有症狀的亨廷頓病患者中,已知左旋多巴可以使舞蹈樣動作增加,左旋多巴可引起舞蹈樣動作的患者比不引起舞蹈樣動作者更可能發生本病,可以誘發處於亞臨床狀態的患者出現臨床表現,用於早期診斷,該試驗存在一定的假陰性反應,陰性結果不能完全除外發病的可能性。PET檢查發現尾狀核部位的葡萄糖代謝減低,也可以出現在亞臨床狀態的患者,可用作超早期診斷。在亞臨床患者如果基因檢查發現亨廷素基因(TT15)三核苷酸串聯重複序列異常擴展超過40可以進一步確定診斷。由於亨廷頓病具有完全外顯的常染色體顯性遺傳特點,因此亨廷頓病的早期基因診斷具有重要意義,為產前診斷和遺傳諮詢提供可靠的依據。[4]
鑑別診斷
多數亨廷頓病患者有家族史,但通過基因檢查手段也發現一些散發患者,所以需與其他類型的遺傳性和散發性舞蹈病進行鑑別。在家族性疾病中齒狀核-紅核-蒼白球-丘腦下核萎縮、良性遺傳性舞蹈病和家族性棘紅細胞增增多增多症具有類似的臨床特點。散發性舞蹈病主要包括藥物性、妊娠性、血管疾病、甲狀腺功能亢進型、系統性紅斑狼瘡、狼瘡抗凝固綜合徵、紅細胞增多症、艾滋病和風濕性舞蹈病。對患者進行詳細的臨床檢查和必要的輔助檢查有助於亨廷頓病的鑑別診斷。
1、 良性家族性舞蹈症
良性家族性舞蹈症是一種常染色體顯性、隱性和性連鎖的中樞神經系統疾病,分為嬰兒早期、兒童期和少年早期三種類型,典型臨床症狀為非進行性的舞蹈表現,和亨廷頓病不同之處在於智能和精神均正常,影像學檢查均無明顯異常改變,基因檢查發現早期發病者的基因位於常染色體14p可以採用多巴胺受體拮抗藥進行治療,近來此病是否為一個獨立的疾病還是一個疾病綜合徵受到疑問。
2、 風濕性舞蹈病
風濕性舞蹈病是一種散發的良性自限性疾病,病理改變主要表現為基底核炎性病變,主要發病時間在5~15歲,11歲後女性較多。起病多有精神異常,而後隱匿出現不自主的運動,多涉及面部,可伴有構音障礙和吞咽困難,不自主運動更為唐突、暴發,跳動樣和抽動樣,與亨廷頓病的舞蹈樣運動、非刻板模式不同,有些兒童出現肌張力低下,痴呆則罕見。首次發病後持續時間不超過6個月,但25%的患者在發病2年後有復發。部分患者可以伴隨出現風濕熱、心肌炎和關節炎,血沉快或抗鏈球菌溶血親「O」滴度可增高。影像學檢查無異常改變。早期可以應用青黴素和激素治療治療,但不能縮短舞蹈病的自然病程。
3、神經棘紅細胞病
神經棘紅細胞病是一種伴隨中樞神經系統和周圍神經損害的隱性遺傳性疾病,其特徵為進行性神經退行性變,伴舞蹈樣動作及棘形紅細胞增增多。根據遺傳方式分為常染色體隱性或顯性遺傳的舞蹈病-棘形紅細胞增增多增多症,以及 X-連鎖Mcleod綜合徵兩種類型。臨床表現與亨廷頓病有許多共同特點。此症多於15~35歲、以肢體和軀幹的舞蹈以及口面運動障礙開始發病,也可以出現肌張力不全和帕金森綜合徵的表現,常合併周圍神經病。運動障礙持續進行導致病殘,於50~70歲死亡。患者可以出現嚴重的行為障礙和情緒改變,但痴呆不明顯。頭顱CT檢查顯示紋狀體萎縮,特別是尾狀核頭部萎縮最明顯。血塗片檢查發現外周血的紅細胞為棘紅細胞。血清肌酸磷酸激酶和乳酸脫氫酶含量可增高。肌電圖和肌肉活檢有神經原性肌萎縮。神經病理檢查和亨廷頓病相似,尾狀核和殼核萎縮,小細胞消失、大神經元保存,但沒有泛素和亨廷素陽性的神經細胞核內包涵體。臨床上,神經棘紅細胞增增多增多增多症與亨廷頓病的區別是:隱性遺傳、無明顯痴呆、有周圍神經病和神經元性肌萎縮、棘紅細胞增增多、病理改變沒有亨廷素陽性的神經細胞核內包涵體。
4、 其他類型的舞蹈病
藥物性遲發性運動障礙出現在精神病患者長期應用精神阻滯藥後,最顯著的動作累及口和舌,但手、腿、軀幹和呼吸肌也可發生舞蹈手足徐動症,智能障礙僅出現在部分患者的晚期。此病的診斷主要依靠長期應用精神阻滯藥的藥物史。妊娠性、血管疾病、甲狀腺功能亢進、系統性紅斑狼瘡、狼瘡抗凝固綜合徵、紅細胞增多症可以出現舞蹈症表現,這些疾病均存在相應的內科表現,注意觀察相關的內科症狀其鑑別診斷不困難。
治療
目前沒有任何藥物可以改變亨廷頓病的自然病程,但可以採取措施改善臨床症狀、減少舞蹈樣動作。治療集中在對心理與神經徵候兩方面的症狀治療,同時進行必要的支持治療。要讓患者及可能得病者樹立信心,相互幫助,建成富有樂觀主義的家庭。
亨廷頓病患者腦內γ-氨酪酸(GABA)減少,膽鹼能活動受抑制,而多巴胺活動過度,可選用對抗多巴胺能藥物或多巴胺受體抑制劑。
1、對抗多巴胺能藥物或多巴胺受體抑制劑
丁酰苯類藥物中的氟哌啶醇和吩噻嗪類藥物氯丙嗪、奮乃靜等是主要治療藥物,可以阻滯多巴胺受體。苯酰胺類藥物如硫必利(泰必利)有抗多巴胺能的作用,3次/d,100mg/次。
2、 提高膽鹼的含量
毒扁豆鹼抑制中樞膽鹼酯酶的活性,阻止膽鹼的降解,可改善舞蹈樣運動。
3、 增加中樞神經系統的γ-氨酪酸(GABA)含量
異煙肼是γ-氨基丁酸轉移酶的抑制劑,可能使中樞的γ-氨酪酸(GABA)含量升高,使有些患者有輕到中度的進步。一般劑量為10~20mg/kg,每一療程用藥時間4個月~1年;同時應用維生素B6效果更好。
4、 痴呆症狀治療
目前還沒有很好的藥物。但精神症狀通過藥物治療可以獲得改善。可用阿米替林、多塞平(多慮平)改善患者的抑鬱症狀。對暴躁和憤怒暴發時可用氟哌啶醇和碳酸鋰聯合治療。
5、 神經細胞移植或胚胎紋狀體組織的移植
尚處於探索之中,是否有效還不能確定。
6、其他治療
可配合應用神經系統促代謝藥物、維生素類和能量合劑等。抗自由基治療、抗氧化和抗細胞興奮毒性治療可能也具有一定的療效。此外加強肢體功能訓練和進行心理治療也可以獲得良好的療效。[5]
預後
亨廷頓病通常持續10~20年,病後15~16年死亡,女性患者病程較長。
視頻
亨廷頓病(醫學視頻)
2分鐘神經科學 - 亨廷頓病
亨廷頓舞蹈症可以治療
參考資料
- ↑ 吳志英. 亨廷頓病的分子發病機制及治療進展. 中國現代神經疾病雜誌. 2009, 9 (3): 238-241.
- ↑ 變異蛋白在血細胞中逐漸積累引發亨廷頓病. 2012-9-22 [07 三月 2020] (中文).
- ↑ 中華醫學會神經病學分會帕金森病及運動障礙學組. 亨廷頓病的診斷與治療指南. 中華神經科雜誌. 2011, 44 (9): 638-641.
- ↑ 莫亞勤; 李麓芸; 盧光琇. 亨廷頓病的基因診斷. 遺傳. 2005, 27 (6): 861-864.
- ↑ 中華醫學會神經病學分會帕金森病及運動障礙學組. 亨廷頓病的診斷與治療指南. 中華神經科雜誌. 2011, 44 (9): 638-641.