血友病檢視原始碼討論檢視歷史

{kind=link}

{kind=link}

{kind=link}

{kind=link}
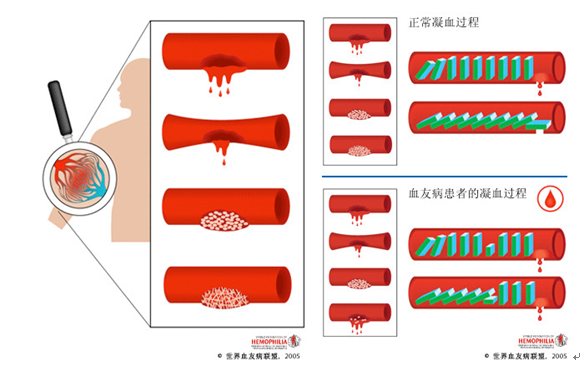
血友病(hemophilia)是一種遺傳性凝血活酶生成障礙引起的出血性疾病。分為血友病A和血友病B兩種。前者為凝血因子Ⅷ(FⅧ)缺乏,後者為凝血因子Ⅸ(FⅨ)缺乏。[1]
血友病的主要病理變化是凝血過程的第一階段——凝血活酶生成障礙。其主要特徵為凝血時間延長,終身具有自發性或輕微損傷後出血傾向。 2018年5月11日,國家衛生健康委員會等5部門聯合制定了《第一批罕見病目錄》,血友病被收錄其中。*[2]
血友病主要是由於遺傳所致,只有基因治療才能根治。目前只能採取預防出血及對症治療。血友病甲嚴重者由於關節腔反覆出血,可造成關節畸形致殘。產前診斷,優生優育對本病發病率的控制尤為重要。
流行病學資料
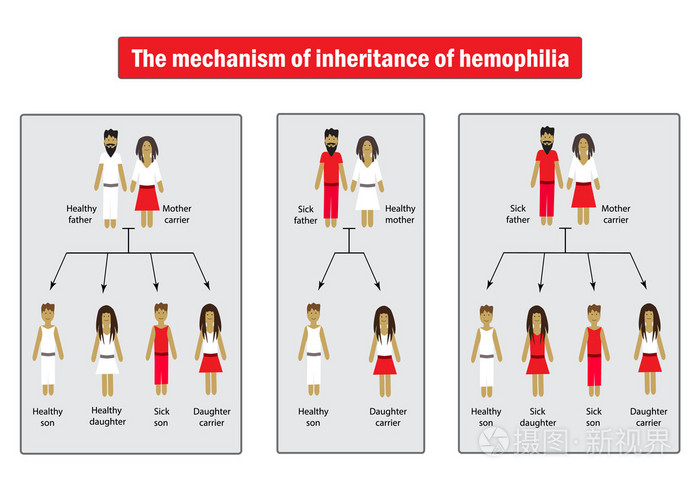
血友病的發病率沒有種族或地區差異。在男性人群中,血友病A的發病率約為1/5000,血友病B的發病率約為1/25000[1]。其中以血友病甲較為多見,占77%,血友病乙約占19.6%。血友病甲缺乏因子Ⅷ,血友病乙缺乏因子Ⅸ,兩者均通過性染色體隱性遺傳,男性發病,女性傳遞,女性傳遞者雖有程度不同的因子Ⅷ或因子Ⅸ活性減低,但一般無出血表現。約有30%的血友病患者家族中無同樣疾病或有關檢查異常發現,可能是由於基因突變所致。[3]
遺傳方式
血友病的遺傳方式為X染色體連鎖隱性遺傳,患病基因位於X染色體。男性的性染色體為XY,若X染色體攜帶血友病基因,不能正常合成FⅧ或 FⅨ,即為血友病患者。女性的性染色體為XX,即使一條X染色體攜帶血友病基因,另一條X染色體仍能合成正常量50%左右的FⅧ或FⅨ,故一般無出血表現,但為血友病攜帶者。血友病患者和攜帶者通過X染色體將疾病遺傳給下一代。
分類
1.血友病A(血友病甲),即因子Ⅷ促凝成分(Ⅷ:C)缺乏症,也稱AGH缺乏症,是一種性聯隱性遺傳疾病,女性傳遞,男性發病。
2.血友病B(血友病乙),即因子Ⅸ缺乏症,又稱PTC缺乏症、凝血活酶成分缺乏症,亦為性聯隱性遺傳,其發病數量較血友病A少。血友病B患者的出血症狀多數較輕。
3.血友病C(血友病丙),即因子Ⅺ(FⅪ)缺乏症,又稱PTA缺乏症、凝血活酶前質缺乏症。為常染色體不完全隱性遺傳,男女均可患病,是一種罕見的血友病。
15~20/10萬男孩中有發病,此發病率在所調查的不同的種族和地域之間沒有差異。發病率以血友病A最多占85%,血友病B占15%,血友病C較少見。
臨床表現
(一)血友病A
1.出血
為本病主要的表現。終身有輕微損傷或手術後長時間出血的傾向。出血程度及發病的早晚與患者血漿中FⅧ活性水平有關。根據出血輕重與血漿中凝血因子活性的水平,將本病分為4型:
(1)重型 血漿中FⅧ活性<1%,常在2歲以前就出血,在嬰兒開始學爬、學走後出現出血症狀,甚至結紮臍帶時出血不止。患者出血部位多且嚴重,常有皮下、肌肉及關節等部位的反覆出血,關節內血腫畸形多見。此外,還可見腎臟出血導致血尿、胃腸道出血、腹腔內出血,肺、胸腔、顱內出血少見。
(2)中間型 FⅧ活性為1%~5%,起病在童年時期以後,以皮下及肌肉出血居多,亦有關節出血,但反覆次數較少,嚴重程度也輕於重型。
(3)輕型 FⅧ活性為5%~25%,出血多在青年期,由於運動、拔牙或外科手術後出血不止而被發現,出血輕微,可以正常生活,參加運動,偶爾發生關節血腫。
(4)亞臨床型 只有大手術後才發生出血,實驗室檢查可以證實為本病,FⅧ活性為25%~40%。
一般而言,凡出血症狀出現越早,病情越重,隨年齡的增長,出血症狀可逐漸減輕,有時可出現無出血症狀的緩解期。出血可在創傷後數小時或數天後發生,也可在創傷或手術後即滲血不止。
2.出血所導致的壓迫症及併發症
出血形成血腫後可導致壓迫症狀:
(1)周圍神經受累 發生率為5%~15%,病人有麻木、劇痛、肌肉萎縮。
(2)上呼吸道梗阻 口腔底部、喉、舌、扁桃體、後咽壁或頸部的嚴重出血甚為危險可引起窒息。
(3)壓迫附近血管 可發生組織壞死。
(二)血友病B
血友病B也可出現類似於血友病A的典型症狀。不同點在於:①血友病B重型患者(FⅨ活性小於2%)較血友病A少,而輕型較多,因此臨床表現較血友病A為輕;
②女性傳遞者也可出血;
③發生抗FⅨ抗體者較少,僅占1%。
(三)血友病C
因子Ⅺ(FⅪ)缺乏症症狀輕,有時僅在手術、拔牙或損傷後出血;其傳遞者一般無臨床症狀,但拔牙後,較正常人容易出血;FⅪ缺乏症常合併其他先天性凝血因子異常,如合併FV、Ⅶ缺乏症。
檢查
1.一般項目
血小板計數正常,束臂試驗陰性,出血時間正常,血塊回縮正常;凝血酶原時間正常,凝血酶時間正常,纖維蛋白原定量正常;凝血時間延長為本病的特徵,但僅在FⅧ:C活性低於1%~2%時才延長,>4%可正常。
2.初篩試驗
凝血酶原消耗試驗(PCT)、活化部分凝血活酶時間(APTT,當因子Ⅷ、Ⅸ的活性減少至正常的30%時,即可延長,可以檢測輕型患者)、簡易凝血活酶生成試驗(STGT)有助於輕型和重型血友病A、B的診斷。
3.確診試驗
可用APTT、STGT、Biggs凝血活酶生成(BiggsTGT)糾正試驗來鑑定血友病類型。如凝血酶原消耗及凝血活酶生成試驗不正常時,可做糾正試驗。正常血漿經硫酸鋇吸附後,尚含有FⅧ、FⅪ;正常血清中含有FⅨ、IXa因子,因此如果患者血漿的部分凝血活酶時間僅被正常硫酸鋇吸附血漿糾正時,為FⅧ缺乏症;僅被正常血清糾正時,為FⅨ缺乏症;如二者皆可糾正,則為FⅪ缺乏症。可將三者加以鑑別。
4.FⅧ、FⅨ、IXa因子活性測定
採用凝血酶原時間一期法,將已知有關因子缺乏的血漿作為基質血漿,加入兔腦浸出液、白陶土懸液、氯化鈣及不同稀釋度血漿或血清後,按凝固時間製成有關因子活性曲線後,對受檢標本進行換算。
5.FⅧRAg的測定
血友病A患者血漿中含量正常或增高。
6.FⅧCAg的測定
在血友病A患者中,血漿Ⅷ:CAg與Ⅷ:C平行減少。
7.VWFAg的測定
血友病A患者正常或增高。
8.基因診斷
血友病分子水平存在着顯著的遺傳異質性,基因診斷血友病是一種有效精確快速的方法,目前主要採用PCR進行基因分析。
9.血友病A攜帶者及胎兒期的診斷和遺傳諮詢
大多數血友病A攜帶者血漿中因子Ⅷ:C的水平僅為正常婦女平均值的50%。近年來,大多數人認為檢測Ⅷ:C與ⅧR:Ag的意義較大,70%~98%的攜帶者比值小於正常。在妊娠第8~12孕周,通過胎兒鏡羊膜穿刺或絨毛取樣,用放射免疫微量法測定ⅧR:Ag及Ⅷ:C,可在產前診斷胎兒是否患血友病,以便考慮中止妊娠問題。近年來基因診斷技術的開展,目前已應用於對傳遞者及產前的檢查。
鑑別診斷
1.血管性假血友病,又稱vonWillebrand氏病,為常染色體顯性遺傳、III型為隱性遺傳,男女均可患病。
家系調查也有助於區別;出血時間延長、阿司匹林耐量試驗陽性,血小板粘附率降低,對瑞斯托黴素血小板無聚集反應,血漿中因子Ⅷ:C/ⅧR:Ag的比例增高或正常,血漿中VW因子缺乏或減少,而血友病A除Ⅷ,C和Ⅷ:C/Ⅷ:Ag比例降低外,其他實驗室檢查均正常。
2.獲得性第Ⅷ因子減少
常見於甲亢、彌散性血管內凝血等疾病,以往無出血史,無家族史,兩性均可發病;臨床伴有原發性疾病症狀和體徵,凝血因子的減少除第Ⅷ因子外尚有其他因子不足,不難與血友病A鑑別。
3.凝血酶複合體減低症
出血症狀及凝血時間與血友病A相似,但患者凝血酶原時間延長,維生素K治療有效。
治療方案
1、 凝血因子替代治療[4]
補充患者缺乏的凝血因子,是目前最常用和最有效的治療和預防出血的方法。[5]
血友病A患者首選FⅧ製劑,包括病毒滅活的人血漿源性FⅧ濃縮劑(凍干人凝血因子Ⅷ)和人基因重組FⅧ製劑[1]。每千克體重輸注1單位 FⅧ可使體內FⅧ:C提高2%,可依此和需要提升的因子水平計算每次的凝血因子輸注量[1]。FⅧ在體內的半衰期為8~12小時,若要使體內FⅧ:C保持在一定水平,可每8~12小時輸注一次[1]。無條件輸注FⅧ製劑者,可選用冷沉澱或新鮮冰凍血漿等。[6]
血友病B患者首選FⅨ製劑。國內沒有人血漿源性FⅨ製劑純品,目前主要採用病毒滅活的血源性凝血酶原複合物(凍干人凝血酶原複合物),也可輸注人基因重組FⅨ製劑 。每千克體重輸注1單位FⅨ製劑可使體內 FⅨ:C提高1%。FⅨ在體內的半衰期約為24小時,故要使體內FⅨ:C保持在一定水平,需每天輸注一次 。無條件輸注FⅨ製劑者,可選用新鮮冰凍血漿等 。
①血友病甲:輸冷沉澱物、新鮮冰凍血漿或因子VIII濃縮物。劑量:輕度關節積血、深部血腫者:因子VIII活性應提高到15%~30%,需輸注10~15U/kg(因子VIII 1個單位相當於正常血漿1ml所含的濃度);嚴重關節積血和深部血腫:因子VIII活性應提高到40%~50%,需輸注15~25U/kg;需作大手術者:因子VIII活性應提高到60%~70%以上,需輸注30~50U/kg。計算公式:需輸注因子VIII劑量(U)=預期達到濃度(U/ml)′40ml/kg′病人體重(kg)。如:要使60kg重的重型血友病甲患者血漿因子VIII活性提高到50%,需輸注因子VIII劑量=0.5U/ml′40ml/kg′60kg=1200(U)。因子VIII在循環中的半衰期約10~12h,應輸注2~3次/d。
②血友病乙:治療原則同血友病甲。輸新鮮冰凍血漿、因子IX濃縮物。劑量:開始劑量40~60U/kg,維持量20U/kg,1次/d(因子IX半衰期較長:20~24/h)。
注意:血友病患者無論是輸注血源性凝血因子還是基因重組的凝血因子,均有可能產生抗體,即凝血因子抑制物 。以血友病A為例,輸注FⅧ製劑後15%~30%的患者會產生抗體 。抗體產生後會中和輸入的凝血因子,降低止血效果 。接受凝血因子輸注的患者應定期檢測抑制物,尤其是當輸注的效果不如從前時 。血友病A患者產生FⅧ抑制物後可換用凍干人凝血酶原複合物治療出血 。
2 其他止血藥物治療
(1)1-去氨基-8-D-精氨酸加壓素(DDAVP):每次劑量一般為0.3μg/kg體重,用50ml生理鹽水稀釋後靜脈滴注,15~30分鐘以上滴完,每12小時1次,1~3天為一療程。該藥多次使用後療效差,如效果不佳時應及時補充FⅧ製劑。此藥主要用於輕型血友病A。少數中間型血友病A可能也有效。DDAVP可致水瀦留等不良反應,幼兒應慎用,2歲以下兒童禁用。
(2)抗纖溶藥物:常用藥物有氨甲環酸、氨基己酸等。口腔出血可含服氨甲環酸。泌尿系統出血時禁忌使用氨甲環酸等抗纖溶藥物。 (3)止痛藥:關節和肌肉出血時可引起疼痛,止痛時禁用阿司匹林和非甾體抗炎藥等影響血小板功能的藥物,可選用對血小板功能無明顯影響的藥物,如對乙酰氨基酚、COX-2抑制劑、曲馬多和嗎啡等。
3 手術治療
關節嚴重畸形,影響正常活動者,在嚴格替代治療情況下,可行矯形手術。
4 局部出血的處理
壓迫止血為主。
5 注意事項
避免外傷和手術,如發生關節出血,應固定患肢。忌服阿斯匹林等影響凝血藥物。
凝血酶原複合物由於含有多種凝血因子,有誘發血栓的風險。凝血酶原複合物應避免抗纖溶藥物同時使用,以免增加血栓風險。[7]
視頻
血友病的形成
血友病遺傳大解密,專家用道具帶你了解血友病遺傳規律
3D醫學視頻凝血與血友病
參考資料
- ↑ 中華醫學會血液學分會血栓與止血學組, 中國血友病協作組. 血友病診斷與治療中國專家共識(2013年版). 中華血液學雜誌. 2013, 34 (5): 461-463.
- ↑ 醫政醫管局. 關於公布第一批罕見病目錄的通知. 2018-06-08 [21 三月 2020] (中文).
- ↑ 中華醫學會血液學分會血栓與止血學組, 中國血友病協作組. 血友病診斷與治療中國專家共識(2013年版). 中華血液學雜誌. 2013, 34 (5): 461-463.
- ↑ 中華醫學會血液學分會血栓與止血學組, 中國血友病協作組. 血友病診斷與治療中國專家共識(2013年版). 中華血液學雜誌. 2013, 34 (5): 461-463.
- ↑ 國家基本藥物臨床應用指南和處方集編委會主編. 國家基本藥物臨床應用指南:2012年版. 北京: 人民衛生出版社. 2013年.
- ↑ 國家基本藥物臨床應用指南和處方集編委會主編. 國家基本藥物臨床應用指南:2012年版. 北京: 人民衛生出版社. 2013年.
- ↑ 國家基本藥物臨床應用指南和處方集編委會主編. 國家基本藥物臨床應用指南:2012年版. 北京: 人民衛生出版社. 2013年.