血友病查看源代码讨论查看历史

{kind=link}

{kind=link}

{kind=link}

{kind=link}
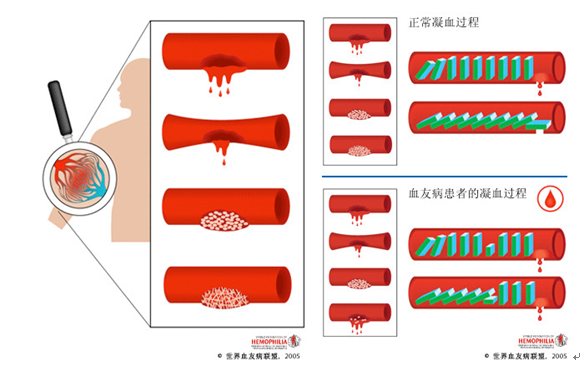
血友病(hemophilia)是一种遗传性凝血活酶生成障碍引起的出血性疾病。分为血友病A和血友病B两种。前者为凝血因子Ⅷ(FⅧ)缺乏,后者为凝血因子Ⅸ(FⅨ)缺乏。[1]
血友病的主要病理变化是凝血过程的第一阶段——凝血活酶生成障碍。其主要特征为凝血时间延长,终身具有自发性或轻微损伤后出血倾向。 2018年5月11日,国家卫生健康委员会等5部门联合制定了《第一批罕见病目录》,血友病被收录其中。*[2]
血友病主要是由于遗传所致,只有基因治疗才能根治。目前只能采取预防出血及对症治疗。血友病甲严重者由于关节腔反复出血,可造成关节畸形致残。产前诊断,优生优育对本病发病率的控制尤为重要。
流行病学资料
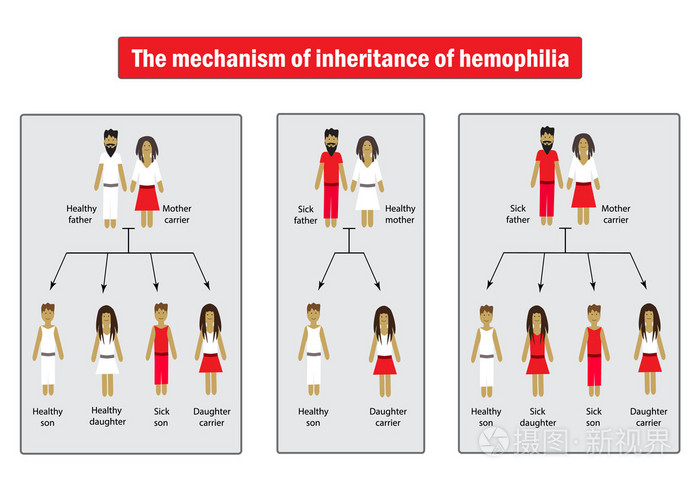
血友病的发病率没有种族或地区差异。在男性人群中,血友病A的发病率约为1/5000,血友病B的发病率约为1/25000[1]。其中以血友病甲较为多见,占77%,血友病乙约占19.6%。血友病甲缺乏因子Ⅷ,血友病乙缺乏因子Ⅸ,两者均通过性染色体隐性遗传,男性发病,女性传递,女性传递者虽有程度不同的因子Ⅷ或因子Ⅸ活性减低,但一般无出血表现。约有30%的血友病患者家族中无同样疾病或有关检查异常发现,可能是由于基因突变所致。[3]
遗传方式
血友病的遗传方式为X染色体连锁隐性遗传,患病基因位于X染色体。男性的性染色体为XY,若X染色体携带血友病基因,不能正常合成FⅧ或 FⅨ,即为血友病患者。女性的性染色体为XX,即使一条X染色体携带血友病基因,另一条X染色体仍能合成正常量50%左右的FⅧ或FⅨ,故一般无出血表现,但为血友病携带者。血友病患者和携带者通过X染色体将疾病遗传给下一代。
分类
1.血友病A(血友病甲),即因子Ⅷ促凝成分(Ⅷ:C)缺乏症,也称AGH缺乏症,是一种性联隐性遗传疾病,女性传递,男性发病。
2.血友病B(血友病乙),即因子Ⅸ缺乏症,又称PTC缺乏症、凝血活酶成分缺乏症,亦为性联隐性遗传,其发病数量较血友病A少。血友病B患者的出血症状多数较轻。
3.血友病C(血友病丙),即因子Ⅺ(FⅪ)缺乏症,又称PTA缺乏症、凝血活酶前质缺乏症。为常染色体不完全隐性遗传,男女均可患病,是一种罕见的血友病。
15~20/10万男孩中有发病,此发病率在所调查的不同的种族和地域之间没有差异。发病率以血友病A最多占85%,血友病B占15%,血友病C较少见。
临床表现
(一)血友病A
1.出血
为本病主要的表现。终身有轻微损伤或手术后长时间出血的倾向。出血程度及发病的早晚与患者血浆中FⅧ活性水平有关。根据出血轻重与血浆中凝血因子活性的水平,将本病分为4型:
(1)重型 血浆中FⅧ活性<1%,常在2岁以前就出血,在婴儿开始学爬、学走后出现出血症状,甚至结扎脐带时出血不止。患者出血部位多且严重,常有皮下、肌肉及关节等部位的反复出血,关节内血肿畸形多见。此外,还可见肾脏出血导致血尿、胃肠道出血、腹腔内出血,肺、胸腔、颅内出血少见。
(2)中间型 FⅧ活性为1%~5%,起病在童年时期以后,以皮下及肌肉出血居多,亦有关节出血,但反复次数较少,严重程度也轻于重型。
(3)轻型 FⅧ活性为5%~25%,出血多在青年期,由于运动、拔牙或外科手术后出血不止而被发现,出血轻微,可以正常生活,参加运动,偶尔发生关节血肿。
(4)亚临床型 只有大手术后才发生出血,实验室检查可以证实为本病,FⅧ活性为25%~40%。
一般而言,凡出血症状出现越早,病情越重,随年龄的增长,出血症状可逐渐减轻,有时可出现无出血症状的缓解期。出血可在创伤后数小时或数天后发生,也可在创伤或手术后即渗血不止。
2.出血所导致的压迫症及并发症
出血形成血肿后可导致压迫症状:
(1)周围神经受累 发生率为5%~15%,病人有麻木、剧痛、肌肉萎缩。
(2)上呼吸道梗阻 口腔底部、喉、舌、扁桃体、后咽壁或颈部的严重出血甚为危险可引起窒息。
(3)压迫附近血管 可发生组织坏死。
(二)血友病B
血友病B也可出现类似于血友病A的典型症状。不同点在于:①血友病B重型患者(FⅨ活性小于2%)较血友病A少,而轻型较多,因此临床表现较血友病A为轻;
②女性传递者也可出血;
③发生抗FⅨ抗体者较少,仅占1%。
(三)血友病C
因子Ⅺ(FⅪ)缺乏症症状轻,有时仅在手术、拔牙或损伤后出血;其传递者一般无临床症状,但拔牙后,较正常人容易出血;FⅪ缺乏症常合并其他先天性凝血因子异常,如合并FV、Ⅶ缺乏症。
检查
1.一般项目
血小板计数正常,束臂试验阴性,出血时间正常,血块回缩正常;凝血酶原时间正常,凝血酶时间正常,纤维蛋白原定量正常;凝血时间延长为本病的特征,但仅在FⅧ:C活性低于1%~2%时才延长,>4%可正常。
2.初筛试验
凝血酶原消耗试验(PCT)、活化部分凝血活酶时间(APTT,当因子Ⅷ、Ⅸ的活性减少至正常的30%时,即可延长,可以检测轻型患者)、简易凝血活酶生成试验(STGT)有助于轻型和重型血友病A、B的诊断。
3.确诊试验
可用APTT、STGT、Biggs凝血活酶生成(BiggsTGT)纠正试验来鉴定血友病类型。如凝血酶原消耗及凝血活酶生成试验不正常时,可做纠正试验。正常血浆经硫酸钡吸附后,尚含有FⅧ、FⅪ;正常血清中含有FⅨ、IXa因子,因此如果患者血浆的部分凝血活酶时间仅被正常硫酸钡吸附血浆纠正时,为FⅧ缺乏症;仅被正常血清纠正时,为FⅨ缺乏症;如二者皆可纠正,则为FⅪ缺乏症。可将三者加以鉴别。
4.FⅧ、FⅨ、IXa因子活性测定
采用凝血酶原时间一期法,将已知有关因子缺乏的血浆作为基质血浆,加入兔脑浸出液、白陶土悬液、氯化钙及不同稀释度血浆或血清后,按凝固时间制成有关因子活性曲线后,对受检标本进行换算。
5.FⅧRAg的测定
血友病A患者血浆中含量正常或增高。
6.FⅧCAg的测定
在血友病A患者中,血浆Ⅷ:CAg与Ⅷ:C平行减少。
7.VWFAg的测定
血友病A患者正常或增高。
8.基因诊断
血友病分子水平存在着显著的遗传异质性,基因诊断血友病是一种有效精确快速的方法,目前主要采用PCR进行基因分析。
9.血友病A携带者及胎儿期的诊断和遗传咨询
大多数血友病A携带者血浆中因子Ⅷ:C的水平仅为正常妇女平均值的50%。近年来,大多数人认为检测Ⅷ:C与ⅧR:Ag的意义较大,70%~98%的携带者比值小于正常。在妊娠第8~12孕周,通过胎儿镜羊膜穿刺或绒毛取样,用放射免疫微量法测定ⅧR:Ag及Ⅷ:C,可在产前诊断胎儿是否患血友病,以便考虑中止妊娠问题。近年来基因诊断技术的开展,目前已应用于对传递者及产前的检查。
鉴别诊断
1.血管性假血友病,又称vonWillebrand氏病,为常染色体显性遗传、III型为隐性遗传,男女均可患病。
家系调查也有助于区别;出血时间延长、阿司匹林耐量试验阳性,血小板粘附率降低,对瑞斯托霉素血小板无聚集反应,血浆中因子Ⅷ:C/ⅧR:Ag的比例增高或正常,血浆中VW因子缺乏或减少,而血友病A除Ⅷ,C和Ⅷ:C/Ⅷ:Ag比例降低外,其他实验室检查均正常。
2.获得性第Ⅷ因子减少
常见于甲亢、弥散性血管内凝血等疾病,以往无出血史,无家族史,两性均可发病;临床伴有原发性疾病症状和体征,凝血因子的减少除第Ⅷ因子外尚有其他因子不足,不难与血友病A鉴别。
3.凝血酶复合体减低症
出血症状及凝血时间与血友病A相似,但患者凝血酶原时间延长,维生素K治疗有效。
治疗方案
1、 凝血因子替代治疗[4]
补充患者缺乏的凝血因子,是目前最常用和最有效的治疗和预防出血的方法。[5]
血友病A患者首选FⅧ制剂,包括病毒灭活的人血浆源性FⅧ浓缩剂(冻干人凝血因子Ⅷ)和人基因重组FⅧ制剂[1]。每千克体重输注1单位 FⅧ可使体内FⅧ:C提高2%,可依此和需要提升的因子水平计算每次的凝血因子输注量[1]。FⅧ在体内的半衰期为8~12小时,若要使体内FⅧ:C保持在一定水平,可每8~12小时输注一次[1]。无条件输注FⅧ制剂者,可选用冷沉淀或新鲜冰冻血浆等。[6]
血友病B患者首选FⅨ制剂。国内没有人血浆源性FⅨ制剂纯品,目前主要采用病毒灭活的血源性凝血酶原复合物(冻干人凝血酶原复合物),也可输注人基因重组FⅨ制剂 。每千克体重输注1单位FⅨ制剂可使体内 FⅨ:C提高1%。FⅨ在体内的半衰期约为24小时,故要使体内FⅨ:C保持在一定水平,需每天输注一次 。无条件输注FⅨ制剂者,可选用新鲜冰冻血浆等 。
①血友病甲:输冷沉淀物、新鲜冰冻血浆或因子VIII浓缩物。剂量:轻度关节积血、深部血肿者:因子VIII活性应提高到15%~30%,需输注10~15U/kg(因子VIII 1个单位相当于正常血浆1ml所含的浓度);严重关节积血和深部血肿:因子VIII活性应提高到40%~50%,需输注15~25U/kg;需作大手术者:因子VIII活性应提高到60%~70%以上,需输注30~50U/kg。计算公式:需输注因子VIII剂量(U)=预期达到浓度(U/ml)′40ml/kg′病人体重(kg)。如:要使60kg重的重型血友病甲患者血浆因子VIII活性提高到50%,需输注因子VIII剂量=0.5U/ml′40ml/kg′60kg=1200(U)。因子VIII在循环中的半衰期约10~12h,应输注2~3次/d。
②血友病乙:治疗原则同血友病甲。输新鲜冰冻血浆、因子IX浓缩物。剂量:开始剂量40~60U/kg,维持量20U/kg,1次/d(因子IX半衰期较长:20~24/h)。
注意:血友病患者无论是输注血源性凝血因子还是基因重组的凝血因子,均有可能产生抗体,即凝血因子抑制物 。以血友病A为例,输注FⅧ制剂后15%~30%的患者会产生抗体 。抗体产生后会中和输入的凝血因子,降低止血效果 。接受凝血因子输注的患者应定期检测抑制物,尤其是当输注的效果不如从前时 。血友病A患者产生FⅧ抑制物后可换用冻干人凝血酶原复合物治疗出血 。
2 其他止血药物治疗
(1)1-去氨基-8-D-精氨酸加压素(DDAVP):每次剂量一般为0.3μg/kg体重,用50ml生理盐水稀释后静脉滴注,15~30分钟以上滴完,每12小时1次,1~3天为一疗程。该药多次使用后疗效差,如效果不佳时应及时补充FⅧ制剂。此药主要用于轻型血友病A。少数中间型血友病A可能也有效。DDAVP可致水潴留等不良反应,幼儿应慎用,2岁以下儿童禁用。
(2)抗纤溶药物:常用药物有氨甲环酸、氨基己酸等。口腔出血可含服氨甲环酸。泌尿系统出血时禁忌使用氨甲环酸等抗纤溶药物。 (3)止痛药:关节和肌肉出血时可引起疼痛,止痛时禁用阿司匹林和非甾体抗炎药等影响血小板功能的药物,可选用对血小板功能无明显影响的药物,如对乙酰氨基酚、COX-2抑制剂、曲马多和吗啡等。
3 手术治疗
关节严重畸形,影响正常活动者,在严格替代治疗情况下,可行矫形手术。
4 局部出血的处理
压迫止血为主。
5 注意事项
避免外伤和手术,如发生关节出血,应固定患肢。忌服阿斯匹林等影响凝血药物。
凝血酶原复合物由于含有多种凝血因子,有诱发血栓的风险。凝血酶原复合物应避免抗纤溶药物同时使用,以免增加血栓风险。[7]
视频
血友病的形成
血友病遗传大解密,专家用道具带你了解血友病遗传规律
3D医学视频凝血与血友病
参考资料
- ↑ 中华医学会血液学分会血栓与止血学组, 中国血友病协作组. 血友病诊断与治疗中国专家共识(2013年版). 中华血液学杂志. 2013, 34 (5): 461-463.
- ↑ 医政医管局. 关于公布第一批罕见病目录的通知. 2018-06-08 [21 三月 2020] (中文).
- ↑ 中华医学会血液学分会血栓与止血学组, 中国血友病协作组. 血友病诊断与治疗中国专家共识(2013年版). 中华血液学杂志. 2013, 34 (5): 461-463.
- ↑ 中华医学会血液学分会血栓与止血学组, 中国血友病协作组. 血友病诊断与治疗中国专家共识(2013年版). 中华血液学杂志. 2013, 34 (5): 461-463.
- ↑ 国家基本药物临床应用指南和处方集编委会主编. 国家基本药物临床应用指南:2012年版. 北京: 人民卫生出版社. 2013年.
- ↑ 国家基本药物临床应用指南和处方集编委会主编. 国家基本药物临床应用指南:2012年版. 北京: 人民卫生出版社. 2013年.
- ↑ 国家基本药物临床应用指南和处方集编委会主编. 国家基本药物临床应用指南:2012年版. 北京: 人民卫生出版社. 2013年.