硬皮病查看源代码讨论查看历史

硬皮病是一种以皮肤纤维化为主,并累及血管和内脏器官的自身免疫性疾病。本病轻重变异程度很大,其中一部分患者病变呈局限性良性皮损,称为硬皮病(scleroderma),另一部分患者有广泛的皮损,并累及内脏器官,称为弥漫性系统硬化(diffuse systemic sclerosis)。本病在风湿性疾病中仅次于系统性红斑狼疮。可称进行性系统性硬化症(progressive systemic sclerosis,PSS),又称系统性硬化(systemicsclerosis)。[1]
目录
症状体征
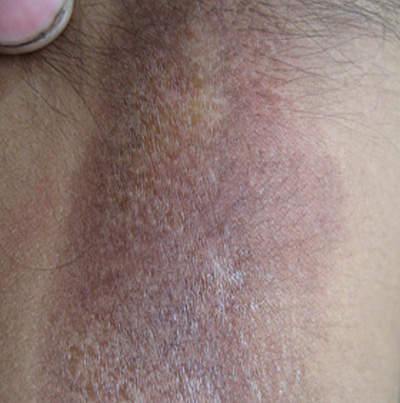
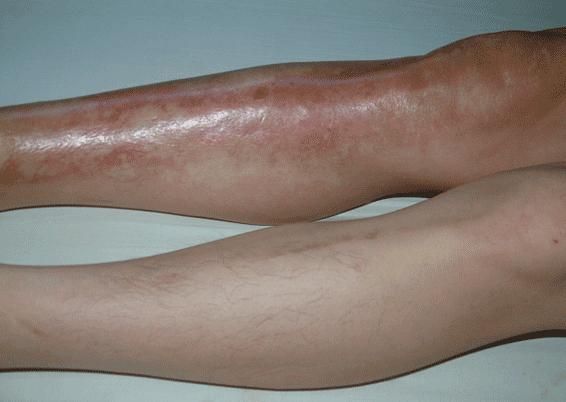
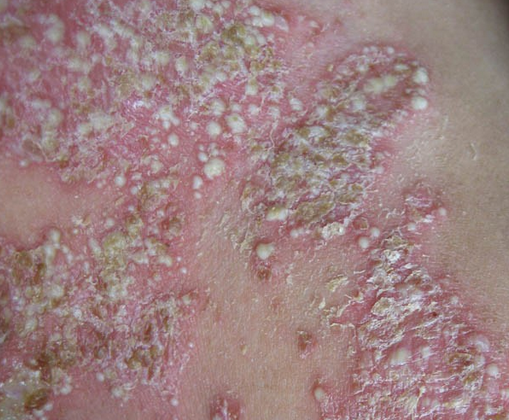
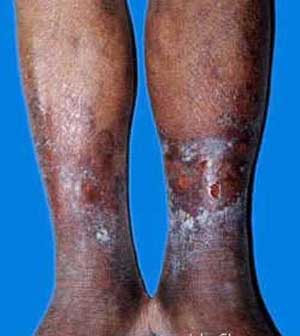
- 肾外表现:患者典型临床表现为手指和面部皮肤增厚,随后躯干和四肢皮肤亦可增厚,常先有雷诺现象,表现为寒冷或情绪紧张诱发血管痉挛,引起手指发白或发绀发作,通常累及双侧手指,有时是足趾,大约80%雷诺现象患者是由于PSS。皮肤改变包括肿胀、红斑、皮肤色素过少或皮肤色素沉着,继而表皮变薄,出现硬结,皮肤紧贴皮下组织。皮肤活检发现表皮萎缩,深层皮肤纤维化,皮肤附件消失,外周血管单核细胞和肥大细胞浸润,尤其是在早期阶段。食管是最常受到累及的内脏器官,继发于食管运动功能不良的咽下困难是常见症状,87%的患者食管测压时发现食管动力学改变,下段食管括约肌压力降低,可导致反流性食管炎和Barrett食管(慢性消化性溃疡及食管炎综合征)。部分患者小肠广泛病变可致吸收不良。大约1/3患者出现肺纤维化导致限制性呼吸功能障碍。30%~50%的患者心脏受累,导致心律失常或进行性心功能衰竭,与动脉粥样硬化性心脏病所不同的是,患者心脏纤维化左心并不多见。
- 肾脏表现:出现临床肾脏病的频率明显少于形态学上的肾脏病变,依临床观察时间的长短不同,估计在1.5%~97%不等。本病患者36%出现蛋白尿,24%出现高血压,19%出现氮质血症,7%出现恶性高血压综合征。在一项报道中,不伴临床肾脏病表现的116例患者,仅有10%在20年随访期内死亡,但伴有蛋白尿、高血压或肾功能不全的94例患者则有60%于同期内死亡。PSS的临床肾脏病表现为:
- 蛋白尿:蛋白尿是常见表现,但很少有肾病综合征表现。早期常为轻度、间歇性或持续性蛋白尿,一般24h尿蛋白定量<500mg,可伴有轻度的镜下血尿及白细胞尿。蛋白尿可以是惟一的表现,但大部分患者同时有轻、中度高血压及慢性肾功能损害,单纯蛋白尿的预后远较同时伴高血压及氮质血症患者好。大约50%的蛋白尿患者与高血压、氮质血症的发生有关。持续存在的蛋白尿提示预后不良,3年内死亡率约64%,而无蛋白尿者仅5%在同期内死亡。
- (高血压:高血压被认为是患者肾受损的一个临床标志,也是肾损害的常见表现,发生率25%~58%,对PSS患者的预后影响较大。合并高血压的患者死亡率比血压正常者高2.5倍,2/3有高血压的患者于20年内死亡。PSS患者的高血压可分为两种类型:一种为慢性高血压,占本病高血压患者的75%~80%,一般起病隐袭,呈慢性经过,为持续性或间歇性,很少发生急性肾功能衰竭。这种良性高血压常发生在病程的晚期,对预后影响不大。只有约15%的慢性高血压患者可导致死亡。另一种为恶性高血压;有些患者在一段时间后,发展为恶性高血压综合征和少尿型肾功能衰竭。有些学者认为恶性高血压综合征通常出现在某些损害肾血流灌往的疾病之后,例如充血性心衰、心包积液、腹部外伤、脱水或感染。Cannon等报道,本病30%的高血压患者表现为恶性高血压综合征,其特点是,血压突然升高,肾功能急剧恶化,最终发展为少尿型肾功能衰竭,常与视网膜病(出血、渗出、视盘水肿)、心衰和脑病并存。该综合征又可称为PSS肾脏危象。尽管危象患者血压急剧上升,但当肾功能继续恶化时,血压通常降至中度高血压水平。80%~90%的恶性高血压综合征患者在发病后2个月内死亡。个别患者恶性高血压综合征可发生在皮肤变硬之前,应于注意。
- 氮质血症:发展为氮质血症的患者预后较差。纽约学者研究表明,血尿素氮超过25mg/dl的患者,死亡率达77%。大多数研究表明PSS并高血压的患者发展为少尿型肾衰时会发生氮质血症,但人们注意到,少数血压不高的患者也可发展为氮质血症。血压正常的PSS患者,氮质血症往往继发于心衰、感染、脱水、外伤或心包积液等,一般不会发展为肾衰。但是,一些有广泛内脏器官受累的病人,氮质血症的发生可能预示着即将发展为少尿症和肾皮质坏死,后者的肾脏病变包括肾小球和血管损害,这些损害与PSS肾脏危象的高血压患者相似。
- 肾脏危象:Traub等总结1955年到1981年期间的临床资料,将PSS肾脏危象定义为:出现高血压或高血压恶化(血压大于160/90mmHg),出现Ⅲ级或Ⅳ级视网膜病变,血浆肾素活性较正常水平增加2倍以上,1个月内肾功能迅速恶化。匹兹堡的一项研究观察了131例PSS肾脏危象患者,观察期限超过33年,其中15例(11%)血压正常。与血压升高者相比,血压正常的肾衰患者表现出更多的微血管病性溶血性贫血和血小板减少性紫癜,有人认为先前使用肾上腺糖皮质激素治疗对其中部分患者可能是一种促发因素。[2]
目前临床将PSS分为以下几种类型:
- 弥漫型:弥漫型PSS可导致多系统病变,病情严重程度、器官分布和疾病进展情况各有不同。典型的弥漫型PSS发病年龄通常比局限型患者要小。起病突然,通常以数周(4~12周)的面部和肢体远端肿胀及雷诺现象为首发症状,伴有疲乏及虚弱等全身症状,皮肤硬化可从四肢远端迅速波及肘部甚至躯干。出现腱摩擦音几乎是弥漫型PSS的独特表现。由于弥漫型PSS发病后数年内可继发严重的多器官损伤(包括肺、心脏、胃肠道和肾脏),因此死亡率较高。弥漫型PSS皮患者临床表现差异很大,并非所有病例都是进行性的,部分患者可能在数年内保持病情稳定。
- 局限型:局限型PSS患者平均年龄大于弥漫型PSS 5~10年,在其他症状出现以前通常有持续多年甚至几十年的雷诺现象。患者肢体远端皮肤增厚,累及指、手、前臂和颜面,肢体病变一般不超过肘部。皮肤增厚通常伴有面部毛细血管扩张。内脏受损较晚,约在起病10年后,部分患者可发生食管运动功能障碍和(或)肺间质损害,肾损害较少。既往称这种以无痛性皮肤症状为主要表现的疾病为CREST综合征,该缩写词于1964年首次用来命名轻度局限型PSS,其临床特点为钙化(calcinosis)多见于手指软组织,雷诺现象(Raynaud phenomenon),食管运动功能障碍(esophageal dysmotility),硬指(sclerodactyly)和毛细血管扩张(telangiectasis)。近期临床观察显示,并非全部患者均具备上述特征,因此这个名称并不完全适用。
- 重叠型:上述任何一型PSS伴有另一种结缔组织病,如PSS与多发性肌炎或皮肌炎或系统性红斑狼疮或舍格伦综合征重叠,称重叠型PSS。仅有PSS内脏损害而无硬皮者不足1%。
发病机制
关于发病机制有四种假说:微血管假说、免疫假说、胶原假说,后病毒病因假说。这几种学说可能结合在一起,通过免疫细胞、血小板、内皮细胞及成纤维细胞产生的细胞因子、生长因子及其他介质组成的网络系统共同发挥作用。
- 微循环假说:本假说认为继发于微血管病变,原发性内皮损伤导致血管痉挛,损伤部位血小板黏附、聚集、活化,发生血管内凝血,内膜细胞增殖,富含黏多糖的物质沉积,导致血管狭窄,局部组织缺血,毛细血管渗透性改变,并通过对邻近间质成纤维细胞的免疫介导,增加胶原沉积,导致组织纤维化。多种血管加压物质被认为是血管不稳定性的介质,包括5-羟色胺(5-HT)、儿茶酚胺、肾素、血栓素A2(TXA2)、前列腺素等。近来认识到局部血管活性物质如内皮源性舒张因子(EDRF)和内皮素在PSS发病机制中的作用。已证明PSS患者的血清内皮素浓度较对照组大约高3倍,受到寒冷刺激后会迅速升高,同时观察到内皮素能促使成纤维细胞有丝分裂,刺激胶原合成。转移生长因子β(TGF-β)也可能参与了PSS的组织纤维化。
- 免疫假说:大量研究表明,免疫反应可能是PSS血管损伤和组织纤维化的起因。临床上PSS可出现自身免疫性疾病的表现,如多发性肌炎、系统性红斑狼疮、舍格伦综合征和原发性胆汁性肝硬化。若将人类口腔癌细胞株IIep-2作为底物,95%~98%的PSS患者可发现ANA阳性。70%~80%的局限型PSS患者有抗着丝点抗体阳性,30%的弥漫型PSS患者和2%伴其他胶原血管病的PSS患者抗Scl-70抗体阳性。其他抗体包括抗SS-A(Ro)抗体、淋巴细胞毒抗体和Ⅳ型胶原基膜抗体等。用敏感的细胞分析方法可在55%~72%的PSS患者中检测到循环免疫复合物(CIC)。CIC的出现与内脏受损有关,尤其是与肺受损有关。50%的PSS患者在硬皮症状出现以前的水肿期可观察到皮肤外周血管的炎症细胞浸润,提示细胞免疫在PSS发病机制中可能发挥作用。这些浸润细胞主要由活化的CD4细胞所构成,肥大细胞也较多见。推测肥大细胞是内皮损伤和纤维化形成中的重要中介,它主要通过释放组胺刺激成纤维细胞增殖和基质合成。据报道,PSS患者具有各种不同类型的细胞免疫异常。目前仍未确定这些免疫异常是原发性抑或继发性,以及是否具有特异性。这些免疫异常包括淋巴细胞对植物血凝素和刀豆蛋白的增殖反应受损,CD8细胞百分率下降,自然杀伤细胞减少,血循环中活化的单核细胞增加。PSS患者未受刺激的外周血单核细胞能产生一种细胞因子,这种细胞因子可促进成纤维细胞生长和胶原合成。PSS患者血清中所含白介素-2(IL-2)的水平较对照组明显增高,其淋巴细胞表达高亲和力的IL-2受体水平更高。对移植物抗宿主反应的研究,给免疫调控参与PSS发病机制提供了强有力的证据。接受骨髓移植的患者生存1~2年后发生的全身性疾病在临床病理和血清学方面的异常都与PSS非常相似。这些患者可发生类似PSS的皮肤改变,并可伴有雷诺现象、干燥面容、多关节痛,以及CIC、ANA和淋巴细胞毒抗体阳性。另外,PSS血管损伤的病理特征与同种移植肾排斥反应非常相似。[3]
- 胶原合成假说:许多研究证实,将PSS患者的单层成纤维细胞进行培养,能够产生多于对照组2~4倍的胶原蛋白和基质多肽,并且能够连续传代。这些增加的胶原蛋白主要是Ⅰ型和Ⅲ型胶原、蛋白多糖、透明质酸和纤维连接素(fibronectin),这些物质的mRNA表达可能通过旁分泌和自分泌途径产生的细胞因子来调节。TGF-β、血小板源性生长因子(PDGF)、IL-1和肿瘤坏死因子α(TNFα)均能刺激成纤维细胞生长,调节纤维母细胞的胶原产物。正常的成纤维细胞暴露于PSS患者的血清后,与正常对照组相比,能产生更多的胶原蛋白。这些发现支持PSS患者的炎症细胞和血小板产生的细胞因子,能诱导成纤维细胞生长和胶原蛋白合成的假说。同时提示PSS患者成纤维细胞的结缔组织合成能力增强不是原发的,可能是上述细胞因子和其他炎症介质相互作用的结果。
- 后病毒病因假说:最近有证据表明,抗同分异构酶抗体能识别与后病毒蛋白同源的某种氨基酸序列。因此提出后病毒病因假说。这一发现可解释在PSS患者家族成员中观察到自身免疫性疾病的高发现象。自身抗原和后病毒蛋白之间的交叉反应为细胞免疫提供了基础,这种细胞免疫直接或间接导致内皮细胞损伤、血管痉挛、凝集反应和组织纤维化。
- 肾损害发病机制:PSS致肾血管损伤的发病机制目前尚不清楚。通过分析目前关于动脉粥样硬化和器官移植中由于严重的内膜增殖而产生的阻塞性血管病的发病机制,本病肾脏硬化症的发病可能涉及到某些免疫紊乱,引起内皮细胞损伤和促有丝分裂原释放,从而导致合成黏多糖和胶原纤维的细胞移行和增殖。肾脏病理学研究提示,这些病变包括增殖阶段和稳定的纤维化阶段。肾血管损伤可以重新修复,这可解释有些PSS患者肾功能衰竭数月以后可停透析治疗,其肾功能可得到自发改善的原因。[4]
目前已清楚,PSS患者的弓形和小叶间动脉损害并不仅仅由于动脉血压升高所致。在许多研究中,死于肾功能衰竭的PSS患者肾脏发生了组织病理学改变,但并未发现血压升高。大量证据表明,肾循环异常伴肾素-血管紧张素系统的活化在肾脏硬化的发病机制中发挥着重要作用:
- PSS高血压患者的肾活检或肾切除标本中经常发现肾血管损伤。
- 伴良性或恶性高血压的PSS患者常有肾皮质血流异常和肾动脉造影的异常。
- 许多PSS患者在恶性高血压发生之前常先有血压增高。已证实这些患者肾素-血管紧张素系统被激活,且几乎所有PSS肾脏危象患者均可出现这种激活。
- 有些伴顽固性高血压和肾素活性增高的PSS患者,双侧肾切除可使其血压恢复正常,少数患者其他症状也得到改善。
- 抑制肾素释放和血管紧张素产生的药物能改善PSS患者的高血压症状,并使少尿症的发生减少。PSS伴恶性高血压和肾功能衰竭的一个不常见特征是其发病突然且迅速进展为少尿症。病理学上表现为双侧肾皮质斑片状坏死。可以假设,肾皮质血管阻塞性内膜损伤与功能性血管收缩相结合(部分是肾素-血管紧张素系统活化的结果),导致这种结果。部分堵塞的肾弓形和小叶间动脉因功能性血管痉挛导致完全.






病理病因
- 本病病因尚不清楚,目前多数认为硬皮病可能是在一定遗传背景基础上再加持久的慢性感染而导致的一种自身免疫性疾病。归纳起来涉及以下几个方面:
- 遗传和环境因素:本病有明显家庭史,可能与HLA-DR3、DR6、C4null等基因有关。
- 结缔组织代谢异常:本病有广泛的结缔组织病变,成纤维细胞培养显示胶原合成的活性明显增高,并发现纤维连接素基因有突变,致结缔组织代谢异常而导致PSS的发生。
- 感染因素:许多患者发病前常有急性感染,如鼻窦炎、咽峡炎、扁桃体炎、肺炎、猩红热等。在患者的横纹肌和肾脏中曾检出副黏病毒样包涵体。
- 细胞因子:很多细胞因子可对胶原成分的表达起调节作用。如TGFβ不仅对细胞外基质有较强的调节作用,还可影响其他细胞因子,包括IL-1β、TNFα1、PDGF、碱性成纤维细胞生长因子等。
- 免疫异常:在PSS患者体内可测出多种自身抗体(如抗核抗体、抗DNA抗体、抗ssR-NA抗体、抗硬皮病皮肤提取液的抗体等),并见B细胞数增多,体液免疫明显增强,CIC阳性率高达50%以上,多数患者有高丙球蛋白血症,部分病例常与系统性红斑狼疮、皮肌炎、类风湿性关节炎、舍格伦综合征等并发而形成重叠综合征。
疾病诊断
应与下列疾病鉴别:
- 局部性硬皮病 其特征为界限清楚,呈线状(线状硬皮病)或斑状(硬斑病)分布,无PSS典型血清学和内脏表现。此病多见于儿童、年轻人、女性。
- 弥漫性筋膜炎伴嗜酸性粒细胞增多:多在不习惯的剧烈运动后发病,多见于青年男性。本病特征为上下肢突然出现压痛性肿胀,肌肉样硬结。面、手、足一般不受累。缺乏雷诺现象和内脏受累。抗核抗体阴性,嗜酸性粒细胞显著增多。组织学特点为深部筋膜、皮下组织及真皮(程度较轻)有广泛性炎症与硬化。
- 化学物、毒物所致硬皮样综合征
- 食用毒性油后先发生急性中毒症状,然后转为慢性病,出现周围神经病、舍格伦综合征、硬皮及内脏损伤。发病是因为变性油所产生的自由基损害血管内皮,不牵涉到免疫机制。长期接触二氧化硅,聚氯乙烯导致的硬皮、雷诺现象等也是毒性反应,因此临床不象典型PSS,无自身抗体。
- 乳房硅酮置入术后所出现的各种结蒂组织病现很受关注。有些患者的临床与抗核抗体检查结果均与典型PSS相似。
检查方法
实验室检查
- 尿液检查:蛋白尿<500mg/24h,镜下血尿及颗粒管型。
- 血液检查:部分病例血中可找到狼疮细胞,血沉正常或轻度升高;蛋白电泳约半数患者丙种球蛋白升高;血BUN、Scr升高,血尿素氮>25mg/dl,Ccr下降;血浆肾素水平升高。
- 免疫学检查
- 类风湿因子(RF):1/3 PSS患者RF阳性。
- ANA:70%PSS患者ANA阳性。
- 抗Scl-70抗体:为弥漫型PSS的标记性抗体,见于50%~60%的患者。
- 抗着丝点抗体:为局限型PSS的标记性抗体,70%~80%的局限型PSS患者抗着丝点抗体阳性。
- 抗核仁抗体:30%~40%PSS患者该抗体阳性,弥漫型PSS阳性率高。
- 抗核糖核蛋白抗体(抗RNP抗体)和抗SS-A(Ro)抗体有时阳性。
其他辅助检查
- 肾活检:肾活检是评价PSS患者肾损害的重要途径。组织学上将肾损害分为急性和慢性两种形式,为评价肾损害的严重性和分型提供了帮助。急性病变(动脉黏液样水肿、小动脉纤维素样坏死、肾小球毛细血管内血栓以及肾梗死)总是见于急性少尿型肾衰,多数提示病情严重。慢性非活动性病变(动脉内膜硬化、肾小球硬化、肾小管萎缩和肾间质纤维化)提示患者为急性肾衰后遗的肾功能损害。轻到中度的慢性病变通常见于病程长、慢性经过或以肾功能不全和轻度蛋白尿伴或不伴轻度高血压为特征的病情稳定者。有些无临床肾病病史的患者,这些非活动性病变仅在尸解时才被发现。
- 肾活检指征:PSS患者肾活检指征有限,而且没有明确定义。对于PSS、狼疮性肾炎和混合性结缔组织病患者的鉴别,肾活检具有相当大的价值。PSS伴急性肾功能衰竭患者,如不能明确诊断为PSS肾脏危象,而且又无法排除间质性肾炎等疾病所致,可通过肾活检而明确诊断。由于PSS患者肾脏病变不能从临床和其他实验室资料来估计,因此肾活检可为PSS的预后提供有用的资料。
- 肾脏病理:PSS患者肾脏病理学改变的实际发生率很难确定,估计在58%~100%。PSS患者肾脏的解剖特征随肾血管损伤的时间长短和严重程度不同而不同。PSS急性肾功能衰竭伴或不伴严重高血压的患者,肾脏大小是正常的,这是因为肾损害发生迅速的缘故。急性肾衰患者肾表面呈细颗粒状或平滑,但在原有慢性病变基础上发生急性肾衰时,肾表面则表现为粗颗粒状。急性肾衰时肾呈鲜红色,肾皮质有大量瘀点、瘀斑和小的楔形出血或淡黄色梗死区。有些病例,皮质梗死病灶非常多且互相融合,使皮质表面呈花斑样。轻度慢性肾功能不全的患者,肾脏有轻微缩小,皮质表面呈细小颗粒状,颜色苍白或呈褐色。
动脉组织学改变
PSS患者肾血管组织学改变的主要病变部位在小叶间动脉和小动脉。血管病变可分为急性和慢性。急性血管病变有以下两种类型:一种为内膜增殖呈黏液水肿样,该类型主要影响较大的动脉。另一种为纤维素样坏死,主要影响小动脉。这些病变可能是局灶性的,也可能是全身性的。
- 急性期:黏液水肿性内膜增殖首先出现在小叶间动脉,少数出现于小弓形动脉,大弓形动脉和叶间动脉常不被累及。血管受累的典型病变为同心圆状黏液水肿性内膜增生,细胞相对少见,管腔变狭窄。内皮细胞水肿,局部脱落,内膜基质松散,苏木精-嗜伊红染色基质呈轻度嗜碱性或透明,三色染色黏液样基质呈透明或淡蓝色。基质中存在少量拉长的肌内膜细胞,这些细胞呈螺旋式同心圆状排列,即所谓“洋葱皮”样结构。有些动脉黏液样基质内含有嗜碱性粒细胞、嗜品红的小簇纤维蛋白样物质,这些物质对纤维蛋白染色呈阳性反应。数量不等的纤维蛋白沉积在狭窄的管腔,狭窄的管腔常充斥淤积的红细胞,碎片红细胞沉积在深层内膜基质,这为微血管病性溶血性贫血参与本病发病机制提供了形态学依据。血管中层变薄扩展,围绕扩张的内膜,血管外径增加,三色染色法显示血管中层轻度硬化。外膜改变不明显,有些可观察到外膜轻度纤维化,可见极少量的单核细胞。PSS患者肾脏超微结构改变报道较少。与光镜下所观察到的黏液样内膜增殖一致,急性小叶间动脉损害的超微结构呈同心圆样线状排列,具有中等电子致密度。小叶间动脉的血管中层变薄,仅有1~2层细胞,其中部分肌纤维萎缩,内质网肿胀,吞噬体增多。外膜超微结构无明显改变。
- 慢性期:慢性动脉损害因管腔致密同心圆状的内膜纤维弹性组织增生而变狭窄,不存在黏液水肿性内膜增生。增生的纤维弹性组织在三色染色下呈深蓝色,说明存在丰富的基质。血管中层一般正常或轻度硬化,具有正常厚度,无急性动脉损伤时所见的扩展现象。外膜改变不明显。超微结构检查可见慢性动脉损害表现为内膜增厚,增厚的内膜由致密的同心圆状的肌内膜细胞层组成,肌内膜细胞被内膜基质构成的致密带、增厚的弹性组织膜以及胶原蛋白束所分隔。血管中层可见轻度增厚的环绕肌纤维的基底膜套。外径50~15μm的小动脉较少出现黏液样内膜增生。PSS小动脉主要表现为纤维素样坏死,既内皮细胞和中层肌纤维肿胀、坏死,强嗜酸性纤维素样物质占据血管腔和内膜,受累血管严重狭窄或堵塞;偶见少量多形核白细胞、淋巴细胞浸润于受损血管内膜或附集于血管壁。此为PSS血管损害的第2种特征性病变。
肾小球
- 急性期:严重PSS患者急性期肾小球损害表现各异。部分小球毛细血管内可见纤维蛋白血栓,呈节段性或小球性分布。这些部位毛细血管内常见红细胞淤积和内皮细胞肿胀变性。偶见轻度毛细血管内或系膜区细胞增多。急性梗死区肾小球出现部分或完全坏死。PSS肾脏危象患者,球旁细胞可出现增生。
- 慢性期:PSS慢性期肾小球病理改变为肾小球硬化,可见血管内凝血、肾小球局部缺血、肾小球毛细血管壁增厚等。电镜观察可见肾小球基底膜增厚,可有轻度、不规则的足突融合。
- 肾小管和间质:PSS患者肾小管和间质的改变取决于血管和肾小球损伤害的分布、严重程度和病程。急性血管损伤时可见皮质坏死灶,出现间质水肿和淋巴细胞浸润,集合管内可见红细胞、白细胞和脱落的小管上皮细胞。在慢性损害部位,可见轻到重度的小管萎缩、少量淋巴细胞浸润和间质纤维化。PSS患者肾小球和血管免疫荧光检查呈多种多样的改变。与组织学改变相似,血管损伤的免疫荧光染色通常是局灶性的。急性损伤时,内膜可见IgM、C3和纤维蛋白原强阳性,也可见IgG、IgA、C1q和C4沉积。慢性血管损伤,可见少量IgM和C3沉积,但无纤维蛋白原沉积。肾小球和肾小管的病变与动脉一样,染色最深、最普遍的是IgM和C3。少数情况下可见IgG、IgA、C1q和C4沉积。
X线检查
系统型患者往往显示牙周膜增宽;食管、胃肠道蠕动消失,下端狭窄,近侧增宽,小肠蠕动减少,近侧小肠扩张,结肠袋呈球形改变;指端骨质吸收;两肺纹理增粗,或见囊状改变;软组织内有钙盐沉积阴影等。
并发症
主要并发症为恶性高血压及肾脏危象。
预后
PSS患者5年存活率在58%~100%。PSS的预后主要取决于内脏血管损伤的范围和程度。多数研究指出,如肾脏受损继而累及心脏、肺的患者存活率显著降低。肾受损是弥漫型PSS最常见的死亡原因之一,即使较轻度的肾脏受损也可使预期寿命缩短。






用药治疗
局限型PSS
小片皮肤损害可用氟化皮质擦剂。坚持体疗和物理治疗,如音频、蜡疗等以改善带状硬皮的肢体关节挛缩,增加肢体动能。口服维生素E,300~1200U/d,有一定效果。
弥漫型PSS
- 去除感染病灶,增加营养,注意保暖,避免过度劳累与精神紧张。
- 抑制胶原增生:常用药物为:
- 青霉胺(D-penicillamine):该药有干扰胶原分子间交联作用,抑制新胶原生物合成。开始250mg/d口服,逐渐增至1.0~1.5g/d,连服2~3年。对皮肤增厚和营养性改变有一定疗效。但应密切注意可能产生的副性反应,如皮疹、肝、肾损害、骨髓抑制等。
- 秋水仙碱(colchicine):能阻止原胶原转化为胶原、抑制胶原积聚。剂量为0.5~1.5mg/d,连服3个月至数年,对皮肤硬化、雷诺征及食管改变有一定疗效。
- 对氨基苯甲酸钾potassium para-aminobenzoate):能增强单胺氧化酶活性,促使过多的5-HT分解,改善皮肤症状,口服3~4g/d,连服数月至数年。
血管扩张剂
对频发的雷诺现象,可试用钙通道阻滞药,如硝苯地平硝苯吡啶等。此类药物能一时性扩张痉挛的血管,疗效有限。对PSS肾脏危象患者,用血管紧张素转换酶抑制药(ACEI)卡托普利(巯甲丙脯酸)治疗,已取得积极效果,不仅能控制肾脏危象,而且对PSS其他症状也有一定疗效。研究提示PSS患者肾素-血管紧张素系统的激活先于肾脏危象的发生,因此,很难将继发于PSS肾损害的高血压和与PSS并存的原发性高血压区分。伴有轻度高血压的PSS患者应检测血浆肾素活性。如果血浆肾素活性增高,应使用ACEI类药物进行抗高血压治疗。如果肾素水平不高或存在ACEI类药物使用的禁忌证,则可使用其他药物如β受体阻滞药、利尿剂、α受体阻滞药、钙通道阻滞药、中枢兴奋药物甲基多巴或可乐定等。但β受体阻滞药有可能加剧雷诺现象,应予以注意。
生物制剂
随着对PSS发病机制的深入研究,某些生物制剂已开始试用于治疗PSS,如基因重组γ干扰素注射剂能向下调节成纤维细胞合成胶原,效力持久,用于临床已收到初步效果。前列腺素E1也有较好疗效并可改善雷诺现象。
糖皮质激素
糖皮质激素可减轻皮肤的炎症水肿、减轻肺动脉高压,但对肾损害无效。
透析治疗和肾移植
虽然上述抗高血压和其他治疗已使PSS肾脏危象患者1年生存率显著提高,但之后仍有大量PSS伴恶性高血压或肾衰的患者发生持续性肾功能恶化,需要长期腹膜透析、血液透析或肾移植。PSS患者大动脉痉挛、皮肤硬化、血管病变致动静脉瘘关闭,使血液透析遇到较多困难,而腹膜纤维化和腹膜血流功能性减少致腹膜清除能力下降又使腹膜透析效果减低,并易发生感染。少数报道表明,有些伴恶性高血压和肾衰的本病患者进行肾移植后,其肾功能在数月至1年自行恢复,尿量增加,不再需要透析。另一项研究表明,20例患者透析治疗3月以后继续使用ACEI,11例患者肾功能得到改善,不再需要透析。这种迟发性病情缓解的原因并不清楚,可能与血管痉挛的减轻、肾血管损伤的修复导致肾灌注、肾小球滤过和肾小管功能得到部分恢复有关。1973年Richardson首次报道1例PSS患者经双侧肾切除治疗高血压后成功地进行了肾移植术,之后在更多的PSS患者也成功地进行了肾移植术。但是累及到肾、肺、心脏或胃肠道的广泛病变则成为这种治疗模式的绝对或相对禁忌证。虽然有报道移植肾中PSS复发者较多见,组织学检查也支持复发的诊断,但这些组织学发现很难与排斥反应或环孢素毒性相鉴别,伴有内皮细胞毒性和血管痉挛特性的环孢素是否增加了同种异体肾移植后PSS复发的可能,或是接受肾移植的PSS患者对
饮食保健

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
食疗方
- 虫草鸡汤:冬虫夏草15—20克、龙眼肉10克、大枣15克、鸡1只。将鸡宰好洗净,除内脏、大枣去核与冬虫夏草和龙眼肉,一起放进瓦锅内,加水适量,文火煮约3小时,调味后食用。功能补脾益肾养肺安神,适用于肺脾肾虚者。
- 田鸡油炖冰糖:田鸡油5克、冰糖适量,放入锅内,加水适量,炖烂即可食用。功可补肾益精,适用于硬皮症患者。
- 沙虫瘦肉汤:沙虫干50克、猪瘦肉200克,洗净后同放入炖盅中,放入适量水,煮沸后改文火炖约一个半小时,加盐适量,饮汤食猪肉。适用于各种慢性虚损性疾病。
- 枸杞甲鱼汤:甲鱼1只、枸杞60克。取甲鱼除去肠脏及头,洗净,放在锅内,加入枸杞,添足清水,用文火慢慢煨熟,添下调味佐料,食甲鱼肉。功能滋阴潜阳,补虚扶正,适用于虚损证患者。
饮食注意事项
如是局限性硬皮病,皮肤病变面积无扩大,病情稳定者,皮肤病变在硬化、萎缩期者,可适当进食温性食物;辩证为湿热瘀阻者,症见皮肤红肿、皮温较高,病变皮肤面积加大,病情发展,皮肤病变在初期或肿胀期者,则不宜进食温性食品,尤其不能进食辣椒、韭菜、酒、羊肉、狗肉等,可以适当进食寒凉性食物。
另外,皮肤硬化严重的硬皮病患者,还可适当进食咸味食物,如海带、紫菜、牡蛎、盐等,中医理论认为咸味食品有软坚散结的作用,可以促进皮肤软化;皮肤肿胀明显者,可以适当多进食山药、薏米、白扁豆、小麦、冬瓜、白茅根等具有健脾化湿、利水作用的食品;百合具有润肺止咳、清心安神功效,现代药理研究证明,其有抑制胶元纤维硬化增生作用,所有硬皮病患者均可服用。
温性食物有温中、补虚、驱寒的作用,主要有羊肉、狗肉、鸡肉、鸽子、麻雀、羊奶、酒、辣椒、姜、胡椒、葱、蒜、韭菜、红糖、核桃等。
寒凉性食物有清热、泻火、解毒、滋阴的作用,主要有黄瓜、西瓜、苦瓜、丝瓜、冬瓜、雪梨、绿豆、豆腐、豆浆、豆豉、白菜、莲藕、甲鱼、银耳、牡蛎、绿茶等。
预防护理
本病病因不明,尚无有效措施预防其发生。对本病患者应早期诊断及积极治疗,防止发生高血压及肾脏危象,努力降低死亡率。
视频
视频